Advances in Microbiology
Vol.
12
No.
01
(
2023
), Article ID:
63439
,
10
pages
10.12677/AMB.2023.121006
隐–荧光DNA扩增
刘自厚1,刘亮伟2,3*
1河南农业大学国际教育学院,河南 郑州
2河南农业大学生命科学学院,河南 郑州
3农业部农业酶工程重点实验室,河南 郑州
收稿日期:2023年3月1日;录用日期:2023年3月25日;发布日期:2023年3月31日

摘要
目的:隐–荧光(Hf: hidden fluorescent) DNA通过带荧光素及10 nt之内淬灭基团的Hf引物扩增。方法:本研究设计Hf_Pf引物带有dT12_Fam荧光素-dT16_BHQ1淬灭基团,扩增5851 bp HfDNA pET28a-xylanase,探讨Hf_Pf引物Tm (退火温度)适合计算器、HfDNA扩增条件、T5 DNA酶(T5exo)切割产物检测、及T5exo酶切动力学。结果:Hf_Pf Tm计算器为Oligo、IDT,经非等量引物PCR优化扩增HfDNA,激光共聚焦定性检测HfDNA酶切产物Fam荧光、酶标仪定量检测荧光值19,683 a.u。根据HfDNA浓度–荧光值方程,T5exo酶促动力学参数Km 0.1 nM,弥补解离常数KD不足。结论:本研究扩增隐–荧光HfDNA,提供了DNA酶学研究新材料。
关键词
隐–荧光DNA,扩增,荧光检测

Amplification of Hidden Fluorescent DNA
Zihou Liu1, Liangwei Liu2,3*
1International Education College of Henan Agricultural University, Zhengzhou Henan
2Life Science College of Henan Agricultural University, Zhengzhou Henan
3The Key Laboratory of Enzyme Engineering of Agricultural Microbiology, Ministry of Agriculture, Zhengzhou Henan
Received: Mar. 1st, 2023; accepted: Mar. 25th, 2023; published: Mar. 31st, 2023

ABSTRACT
Objective: Hidden fluorescent (Hf) DNA substrates can be amplified by Hf primer that has a fluorescein and a quencher within 10 nt. Method: The study designed a 20 nt Hf_Pf having the dT12_Fam and the dT16_BHQ1, amplified a 5851 bp HfDNA pET28a-xylanase, determined suitable analyzers for calculating primer Hf_Pf Tm (melting temperature), optimized PCR to amplify the HfDNA substrates, assayed HfDNA-T5exo digestion product fluorescence. Result: Suitable analyzers were Oligo and IDT, and un-equal PCR was optimized to amplify the HfDNA substrates. Instead of the HfDNA substrates, the HfDNA-T5exo digestion products exhibited dT12_Fam fluorescence in quality under a laser scanning confocal microcopy and a 19,683 a.u fluorescence intensity in quantity under a microreader. According to HfDNA concentration-fluorescence intensity function, T5exo kinetics was determined to have a 0.1 nM Km, fulfilling in-adequate of dissociation constant KD. Conclusion: The study amplified HfDNA substrates, provided a new material for assaying DNase enzyme properties.
Keywords:Hidden Fluorescent DNA, Amplification, Fluorescence Assay

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
DNA酶(DNase)性质检测涉及底物double-stranded (ds) DNA和产物核苷酸,但是二者均无法显色,所以dsDNA标记和产物检测成为关键。放射性同位素标记dsDNA最早用于T5exo (T5 DNA外切酶)和VIII型外切酶 [1] - [6] 。后来,荧光染料PicoGreen、SYBR Green I、BEBO染色dsDNA用于RecBCD外切酶、T7噬菌体gene 6外切酶和DNA聚合酶 [7] [8] [9] [10] 。而后,荧光标记ddNTP用于Sanger终止法测序、分子标记探针用于荧光定量PCR、拓扑异构酶 [7] [8] [11] [12] [13] 。但是,同位素衰变导致误差 [14] ,荧光染料普遍染色DNA而不具有特异性,而且荧光染料与dsDNA分子没有定量关系。PicoGreen染色2.7 kb dsDNA检测VIII型外切酶,需要荧光标记25 bp dsDNA (探针引物与反向互补ssDNA退火)检测解离常数KD [4] 。但是,荧光标记ddNTP、探针引物、分子标记探针均不参与dsDNA扩增 [8] [9] [11] [12] [13] [15] [16] 。受Cy5-ATP标记ds break连接中间物荧光检测 [17] 和PCR高效制备dsDNA启发 [18] ,带荧光基团和10 bp之内淬灭基团的隐–荧光(Hf: hidden fluorescein) dsDNA扩增成为可能。
本研究将“带荧光素和10 nt之内淬灭基团的引物”称为隐–荧光(Hf)引物,但是这种目前只作为探针引物、还没有用于扩增dsDNA的报道。HfDNA扩增需要以下条件:1) Hf引物能够扩增dsDNA。dT碱基5位通过linker标记荧光、淬灭基团(图1),3、4位游离能够与模板链氢键匹配,Hf引物理论上可以扩增dsDNA。2) Tm (退火温度)是DNA扩增关键,需要确定Hf引物Tm值的适用计算器。3) HfDNA中dT_荧光素-dT_淬灭基团间距10 bp之内,理论上没有荧光,经T5exo酶切分开dT_荧光素、dT_淬灭基团,理论上产生荧光。
基于上述理论分析,本研究设计20 nt正向隐–荧光引物Hf_Pf,其dT12位荧光素6-Fam (carboxflurescine) (dT12_Fam绿色荧光需要495 nm激发光、521 nm发射光)、dT16位淬灭基团BHQ1 (Black Hole Quencher) (图2),与22 nt反向引物Pr协同扩增5851 bp的HfDNA pET28a-xylanase (黑曲霉GH11木聚糖酶基因) [19] 。从DNAman、NEB (New England Biolab)、Oligo (上海生物工程公司)、IDT (Integrated DNA Technologies)中确定Tm适合计算器。根据Hf_Pf/Pr Tm差值选用双退火PCR程序 [20] ,HfDNA-T5exo酶切产物经定性和定量检测,进而计算T5exo酶促动力学,为DNA酶研究提供新材料。
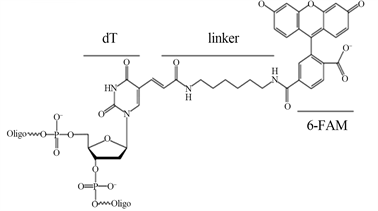
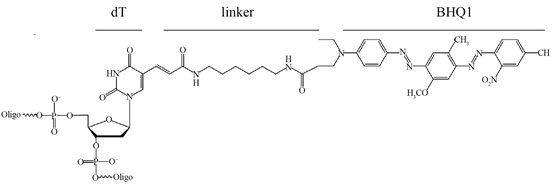
Figure 1. Structure of 6-Fam and BHQ1. dT: nucleotide, linker, 6-Fam: carboxflurescine, BHQ1: Black Hole Quencher
图1. 6-Fam-dT和BHQ1-dT结构式,dT:dT碱基,linker:连接臂,6-Fam:Fam荧光素,BHQ1:BHQ1淬灭基团
![]()
Figure 2. Schematic model of hidden fluorescent HfDNA amplification. Hidden fluorescent primer Hf_Pf has the dT12_Fam fluoresin-the dT16_BHQ1 quencher, Hf_Pf primer has a 10 nt homology arm (HA) with the reverse primer Pr, the 5851 bp HfDNA was amplified by PCR, and was optimized to increase quantity by un-equal primer PC, T5 DNA exonucleae digestion of the HfDNA releases the dT12_Fam from the dT16_BHQ1, and releases fluorescence
图2. 隐–荧光HfDNA扩增模式。隐–荧光引物Hf_Pf带dT12_Fam荧光素-dT16_BHQ1淬灭基团,Hf_Pf与反向引物Pr有10 nt同源(HA),PCR扩增得到全长5851 bp HfDNA,经非等量引物PCR优化提高HfDNA扩增量,HfDNA经T5 DNA酶切分离dT12_Fam与dT16_BHQ1,从而产生荧光
2. 方法与材料
2.1. 材料
Q5 DNA聚合酶、T5 DNA外切酶(T5exo)由NEB (New England BioLabs,中国)公司提供,Hf_Pf引物(dT12_6-Fam, dT16_BHQ1)和常规引物Pr由上海生物工程公司合成(表1)。Hf引物稀释和保存与常规引物相同,干粉在−20℃下保存1年,荧光素不稳定需要避光保存。质粒pET28a-xylanase为本实验室构建 [19] 。
2.2. HfDNA扩增
PCR体系加入54 ng pET28a-xylanase质粒,250 μmol Hf_Pf,250 μmol Pr引物,200 μM dNTPs,1 U Q5 DNA聚合酶,1 × Q5 DNA聚合酶缓冲液,以水补足50 μL体系。PCR程序为:98℃预变性3 min,98℃变性30 s,双退火程序(69℃退火15 s,57℃退火15 s),72℃延伸3 min 40 s,30个循环,72℃延伸10 min。扩增dsDNA用Thermal Cycler Block 5020热循环仪(Thermo Scientific, USA),在1%的琼脂糖凝胶中电泳(DYY-5型电泳仪),EB染色后在302 nm波长下照像,电泳分析软件Gel-Pro Analyzer 4.0 (Media Cybernetics, USA)。
非等量引物扩增HfDNA:引物Hf_Pf与Pr有10 bp同源,容易形成引物二聚体,将Hf_Pf和Pr引物量分别降低20%,探讨非等量引物对HfDNA扩增量影响。
酶切产物定性、定量分析、酶促动力学需要大量HfDNA,利用大量–高纯–高浓度DNA方法制备HfDNA [21] 。将40管(共2000 μL)PCR产物经CsCl-EB超速离心后,以DNA纯化柱回收HfDNA。质粒DNA、HfDNA浓度用Nanodrop 2000检测(Thermo Scientific, USA)。
2.3. HfDNA用于T5exo酶活性检测
T5exo酶切反应:加入600 ng HfDNA,2 μL (10 U/μL) T5exo、1 × T5exo buffer,以水补足10 μL反应体系,在37℃酶切30 min,每个反应设置3个平行,阴性对照加入灭活的T5exo (加入10% SDS经70℃孵育30 min),以水为空白对照。
酶切产物dT12_Fam定量检测:HfDNA-T5exo反应液以H2O补足150 μL,加入黑色酶标板,选择520~550 nm波段用SpectraMax® i3x酶标仪检测(Molecular Devices, Thermo Fisher Scientific, USA),以动态方式(dynamics)每90 s采集一次数据,采集21次荧光值平均值,水为空白对照。
用IBM SPSS Statistics软件分析数据显著性,依次选用“分析”→“比较均值”→“单因素ANOVA”,“两两比较”:选择“LSD (L)、Tukey s-b (K)和Waller-Duncan”参数(Duncan’s multiple range test, P < 0.05),“选项”:描述、方差齐性检验,区分显著性差异。
酶切产物Fam定性检测:从HfDNA-T5exo酶切产物中取出5 μL样品滴加到载玻片上,镊子夹住盖玻片倾斜覆盖液体,避免气泡产生,用A1R HD25激光共聚焦显微镜在488 nm激光下检测(Nikon Corporation, Japan)。
2.4. HfDNA用于T5exo酶促动力学
构建荧光值-HfDNA浓度方程:酶切体系如上,只是150 μL体系分别加入50、100、300、500 ng HfDNA,加入黑色酶标板,以SpectraMax® i3x酶标仪实时检测37℃反应30 min荧光值,以动态方式每90 s检测一次荧光值,共采集21次数据平均值。
酶促动力学检测:150 μL反应体系分别加入50、100、300、500 ng HfDNA,1 U T5exo,加入黑色酶标板。实时检测37℃反应30 min荧光值,每个反应3次重复,根据荧光值–时间变化趋势,取前7 min反应数据线性拟合,根据拟合方程计算各时刻荧光值,从而减小荧光值波动的误差。根据标准曲线计算反应速度(nM/s),与底物(nM)进行Hill方程拟合,得到最大反应速度Vmax和亲合常数Km。
3. 结果与分析
3.1. HfDNA扩增
如图2所示,Hf_Pf引物带dT12_Fam荧光素和4 nt之内dT16_BHQ1淬灭基团,BHQ1淬灭10 nt内荧光,Hf_Pf引物理论上无荧光。以Hf_Pf及Pr引物、质粒为模板通过反向PCR扩增5851 bp 隐–荧光HfDNA。HfDNA中dT12_Fam和dT16_BHQ1间距4 bp,HfDNA理论上无荧光。经T5exo酶切后,dT12_Fam与dT16_BHQ1分离,dT12_Fam理论上产生绿色荧光,通过激光共聚焦显微镜和酶标仪定性、定量检测。
Hf_Pf Tm值计算是扩增HfDNA的关键。分别用DNAman、NEB、Oligo、IDT计算Hf_Pf Tm值,根据能否扩增出HfDNA确定适合计算器。由表1可知,引物Hf_Pf与Pr Tm差值11.3℃~16℃,双退火程序才能扩增HfDNA [20] 。DNAman计算Tm值分别为63/78℃,PCR无目的条带(图3_1)。NEB计算Tm值分别为66℃/79℃,60℃/69℃PCR无目的条带(图3_4)。说明DNAman和NEB不适合Tm计算。

Table 1. Tm values calculated by four analyzers
表1. 引物Tm四种计算值
注:隐–荧光引物Hf_Pf dT12标记荧光素6-Fam,dT16标记淬灭基团BHQ1,Hf_Pf与常规引物Pr 10 nt同源(下划线表示),DNAman:DNAman计算器,Oligo (上海生物工程公司) (https://www.sangon.com/baseCalcuLator),IDT (Integrated DNA Technologies IDT DNA (https://sg.idtdna.com/calc/analyzer) (需注册),NEB (New England Biolab) (http://tmcalculator.neb.com/#!/main)。其中,S/D:以Fam和BHQ1中的单一、双基团计算Tm。
Oligo计算Tm值分别为56.4℃/69℃,57℃/69℃PCR扩增得到目的条带(图3_3)。IDT计算Tm值分别为56℃/67.3℃,57℃/69℃PCR扩增得到目的条带(图3_6)。说明Oligo和IDT适合计算Tm。Oligo计算Tm时考虑修饰基团(Fam和BHQ1)位置影响(位于引物5’端、3’端、中间),加入单一、两种基团时引物Tm分别为56℃、56.1℃ (表1),表明修饰基团降低引物Tm值0.3℃~0.4℃。
![]()
Figure 3. Electrophoresis of HfDNAs. M: λ-DNA/HindIII marker, 1: no HfDNA from PCR using 63˚C/78˚C of DNAman, 2: plasmid pET28a-xylanase control, 3: HfDNA from PCR using 57˚C/69˚C of Oligo, 4: no HfDNA from PCR set by Tm 60˚C/69˚C, 5: HfDNA from optimized PCR, 6: HfDNA from PCR using 57˚C/69˚C of IDT. HfDNA from optimized PCR (lane 7~12). 7: with 200:250 μmol primer Hf_Pf/Pr (121 ng HfDNA). 8: with 250:250 umol Hf_Pf/Pr (149 ng HfDNA), 9: with 200:250 μmol Hf_Pf/Pr (163 ng HfDNA), 10: with 200:250 μmol Hf_Pf/Pr and an 80% plasmid templates (213 ng HfDNA), 11~12: 200:300 μmol Hf_Pf/Pr (119 ng HfDNA)
图3. HfDNA电泳。M:λ-DNA/HindIII marker,1:DNAman Tm值63℃/78℃无HfDNA条带,2:pET28a-xylanase质粒对照,3:Oligo Tm值57℃/69℃PCR得到HfDNA条带,4:NEB Tm值60℃/69℃PCR无HfDNA条带,5:PCR优化HfDNA条带,6:IDT 计算Tm值57℃/69℃PCR得到HfDNA条带。PCR优化扩增HfDNA (Lane: 7~12), 7:200:250 μmol Hf_Pf/Pr (121 ng HfDNA),8:250:250 umol Hf_Pf/Pr (149 ng HfDNA),9:200:250 μmol Hf_Pf/Pr (163 ng HfDNA),10:200:250 μmol Hf_Pf/Pr及80%模板量(213 ng HfDNA),11~12:200:300 μmol Hf_Pf/Pr (119 ng HfDNA)
3.2. 非等量引物扩增HfDNA
Oligo和IDT虽然适合Tm值计算(57℃/69℃),但是Hf_Pf与Pr 10 nt同源导致HfDNA扩增量不足(表1),改变单侧引物量的非等量引物PCR才能提高HfDNA扩增量。由Gel-pro analyzer软件计算Hf_Pf/Pr等量时HfDNA量149 ng为参照(图3_8,表2),Hf_Pf降低20%,HFDNA量减少19% (图3_7)。Pr提高20%时,HfDNA量减少20% (图3_11,12)。Pr降低20%时,HfDNA量增加9% (图3_9)。Pr降低20%且模板量降低20%时HfDNA量增加43% (图3_10,5)。降低单侧引物量可以减少引物二聚体形成 [22] ,多余一侧引物与模板结合延伸得到目的DNA。因为至少3次PCR循环才能产生目的DNA,并作为后续指数扩增的模板,初始模板量过大会减少目的DNA扩增量。
Table 2. HfDNA amplified by un-equal PCR
表2. 非等量引物PCR扩增HfDNA
不带荧光–淬灭基团的同源引物,降低任何单侧引物量10%均可提高DNA扩增量 [20] 。但是,Hf引物却明显不同,降低Hf_Pf量均减少HfDNA扩增量,与Oligo引入修饰基团降低Tm值一致,说明修饰基团降低Hf_Pf引物–模板亲合力,所以HfDNA扩增时只能降低常规引物20%。
PCR优化后大量扩增HfDNA,将40管2000 μL PCR产物经大批量–高纯度–高浓度纯化方法:CsCl-EB超速离心-DNA纯化柱回收。共得到28,715 ng纯化HfDNA,浓度195.5 ng/μL,A260/280可达1.89,A260/230可达2.26。
3.3. HfDNA-T5exo酶切产物检测
Hf_Pf引物dT12_Fam与dT16_BHQ1间距4 nt,HfDNA中二者间距4 bp,所以理论上无荧光,激光共聚焦显微镜检测证实无荧光(图4_2)。600 ng HfDNA经T5exo酶切将dT12_Fam基团与dT16_BHQ1分开 [15] ,理论上产生荧光,激光共聚焦显微镜在488 nm激发光下检测到Fam绿色荧光(图4_1)。
与定性检测不同,酶标仪在520~550 nm波段可以定量检测荧光强度。600 ng HfDNA酶切产物荧光值19,683 a.u (图4_3),阴性对照(T5exo-B)荧光值与水相同(图4_3)。HfDNA-T5exo酶切产物产生Fam荧光,表明HfDNA可以定性、定量检测DNA酶活性。
3.4. HfDNA用于T5exo酶促动力学
首先检测50、100、300、500 ng HfDNA完全降解产物荧光值,得到Fam荧光值-HfDNA浓度方程(图5左):dT12_Fam = 22,225 × HfDNA (Fam指反应后荧光值,反应体系中HfDNA浓度nM,R2 = 0.98)。根据方程,600 ng HfDNA酶切产物荧光值19,683 a.u相当于19,683 ÷ 22,225 = 0.89 nM (图4_3)。600 ng HfDNA理论浓度600 ng ÷ (5,851 bp × 2 × 330) ÷ (0.15 µL ÷ 1000) = 1.0 nM,表明荧光计算值与理论值相近。
![]()
Figure 4. Assay of HfDNA-T5exo digestion product green dT12_Fam fluorescence. Fluorescence dT12_Fam of the HfDNA-T5exo digestion 20× observed by a laser scanning con-focal microscope ①, with the HfDNA pET28a-xylanase as the negative control ②. dT12_Fam fluorescence intensity of the HfDNA-T5exo digestion assayed for 19,683 a.u by a Microplate Reader ③, with water as the blank control (H2O), and the negative control (T5exo-B). The groups a and b were significantly different (Duncan’s multiple range test, P < 0.05)
图4. HfDNA-T5exo酶切产物Fam荧光检测。激光共聚焦显微镜检测HfDNA-T5exo酶切产物dT12_Fam荧光20× ①,HfDNA pET28a-xylanase无荧光②。酶标仪定量检测HfDNA-T5exo酶切产物Fam荧光值19,683 a.u ③,阴性反应荧光值(T5exo-B)与水(H2O)相同,a和b具有显著性差异(Duncan’s multiple range test, P < 0.05)
为探讨HfDNA适用性,检测T5exo酶切HfDNA酶促动力学。在150 µL体系下37℃ 30 min酶切50、100、300、500 ng底物,每90 s实时检测荧光值。取不同底物浓度下6 min (6 × 90 s)荧光值线性拟合避免误差 [23] ,则底物反应速率(nM/s) = 线性方程斜率 × 540 s ÷ 22,225 (标准曲线斜率) ÷ 540 s,与HfDNA底物浓度拟合Hill方程(图5右),得到Vmax为8.1 × 10−4 nM/s、亲合常数Km为0.1 nM值。则1 min内产生核苷酸 = 8.1 × 10−4 nM/s × 60s = 0.486 nM,接近于1U的T5exo定义(0.5 nM/min dsDNA降解产物)。T5exo酶切速率 = 8.1 × 10−4 (nM/s) × 2 (dsDNA) = 16.2 × 10−4 nM/s*nt/U,说明T5exo有较强持续外切活性。亲合常数Km为0.1 nM,弥补了KD值不足 [1] [5] ,结果表明HfDNA可以表征DNA酶活性。
![]()
Figure 5. Standard curve (left) and kinetics of T5 exonuclease digestion of HfDNA (right). The insert was change of dT12_Fam fluorescence along with time of standard line using 20 U T5exo and kinetics using 1 U T5exo respectively
图5. 标准曲线(左)和T5exo酶切HfDNA动力学(右)。插图分别是标准曲线时20U T5exo-HfDNA荧光值–时间线性拟合,酶促动力学时1U T5exo-HfDNA荧光值–时间线性拟合
基于放射性标记DNA的T5exo转换常数18 nt/s [1] ,则T5exo浓度(说明书无浓度) = 16.2 × 10−4/18 = 0.9 × 10−4 nM/U = 0.9 × 10−4 × 291 (Aa) × 110 (Aa分子量) ng/U = 2.9 ng/U,又因为1 U = 0.1 µL,所以,T5exo浓度 = 29 ng/µL。T5exo转换常数18 nt/s,接近于持续外切活性较高的VIII外切酶Kcat 18.8/s [4] ,高于λ外切酶(λ exonuclease) Kcat 9.3/s [24] 。
4. 讨论
本研究以隐–荧光引物Hf_Pf扩增带有dT12_Fam和dT16_BHQ1的5851 bp HfDNA、经非等量引物PCR优化,HfDNA-T5exo酶切产物定性、定量检测。据笔者所知,还没有Hf引物扩增HfDNA的报道。5851 bp长链HfDNA扩增基于:1) 接近放射性标记40 kb T7噬菌体DNA、2.7 kb质粒pUC19 DNA [1] 、荧光染料PicoGreen染色2.7 kb线性pUC19 DNA [4] 。2) 5851 bp HfDNA能够扩增,则较短HfDNA更容易扩增。3) 针对T5exo Kcat 18.1/s,5851 bp HfDNA底物更好显示T5exo持续外切活性。淬灭基团导致5~7 nm荧光共振能量转移(fluorescence resonance energy transfer: FRET)从而不激发荧光 [25] ,一个DNA螺旋10 bp间距3.4 nm,所以Hf引物需要荧光–淬灭基团间距10 nt之内,间距4 nt的dT12_Fam和dT16_BHQ1的Hf_Pf引物和HfDNA无荧光,T5exo酶切分离dT12_Fam和dT16_BHQ1产生荧光,经激光共聚焦和酶标仪定性和定量检测。化学反应连接荧光素-DNA时出现连接效率问题 [26] [27] [28] ,PCR扩增得到每个DNA分子带一个隐–荧光分子,淬灭基团10 nt之内引入第二个荧光基团可以增强荧光强度。
HfDNA特异性问题。相比而言,放射性同位素标记dsDNA制备复杂 [1] [3] [4] ,需要加入放射性标记的3H、32P原子,利用胞内dsDNA系统合成,而且放射性同位素危害人体健康。以HfDNA得到T5exo亲和常数Km 0.1 nM,弥补了放射性同位素标记dsDNA无法检测Km值的不足 [1] 。以酸可溶性(acid-soluble)方式检测酶切产物无法得到T5exo亲合常数Km [1] 。检测VIII型外切酶活性时 [4] ,通过5’端dT_Fam荧光素标记25 bp dsDNA (5’-[FluorT] AGAGCTTAATTGCTGAATCTGGTG-3’退火反向互补ssDNA),只能得到RecE564解离常数KD 70 nM,不同于PicoGreen染色2.7 kb dsDNA酶切底物,λ exonuclease得到KD 161 nM。与本研究以HfDNA得到T5exo Km 0.1 nM相比,相差近3个数量级,说明KD表示底物亲合力有较大误差,因为T5exo (Kcat 18.1/s )~1 s即可完全降解分子标记25 bp dsDNA,无法表征酶切2.7 kb pUC19 dsDNA底物亲合力,另外,荧光燃料PicoGreen对ssDNA和dsDNA均染色,ssDNA与dsDNA结合的染料分子多少不一致,而且不能定量dsDNA结合多少个染料分子。
荧光值单位问题。因为不同设备荧光单位不一致,荧光值均以绝对单位表示(a.u: arbitrary unit) [26] [27] [28] [29] 。Fam荧光值-HfDNA浓度标准曲线斜率22,225以150 µL检测,而Cy5荧光-Cy5DNA标准曲线斜率29,079以200 µL检测 [30] ,表明200 µL检测体系Cy5比150 µL Fam荧光强度高,增大检测体系可以提高荧光检测准确度。基于相对荧光单位概念,Hf-DNA-Fam荧光方程斜率22,225 a.u除以检测体系150 μL得到Fam相对值荧光强度148.2 a.u /μL。Cy5DNA-Cy5荧光方程斜率29,079 a.u除以200 μL检测体系得到Cy5相对值荧光强度145.4 a.u /μL,则二者相近。表明以检测体系的相对值荧光强度浓度标准曲线可以用于不同荧光素。将荧光值绝对单位19,683 a.u除以HfDNA浓度195.5 ng/µL得到相对荧光强度100.7 a.u/(ng/µL),可以排除HfDNA浓度的影响,则荧光值之间平行性更好。与水作为空白对照相比,阴性对照中T5exo变性影响荧光值释放,从而显示水作为空白更合理。
Tm值计算问题。DNAman、NEB计算Tm值时与理论公式4 × (G + C) + 2 × (A + T)、经验公式62.3 + 0.41 × (GC%)相同。长片段引物(megaprimer)构建重组质粒时 [31] [32] 、引物与模板匹配的3’端序列用于计算Tm [32] ,这些计算公式 [33] 根据氢键数目的多小、或GC%含量计算Tm值,只能计算常规引物Tm值 [34] ,不适合计算Hf引物Tm值。Oligo和IDT适合Hf引物Tm值计算,Hf引物Tm值比前者低6℃~10℃,加入修饰基团后Tm值进一步降低0.3℃~0.4℃。这是因为:1) 荧光–淬灭基团通过较长linker连接dT (图1),2) Oligo和IDT用最近–邻(nearest-neighbor)方法得到碱基平均热动力参数计算Tm值 [35] 。Oligo只是平均考虑修饰基团的5’、3’、及中间位置影响,实际上修饰基团越近3’端对Tm影响越大,3) Q5 DNA聚合酶融合了Sso7d DNA结合域,提高DNA聚合速度、持续合成能力、DNA保真度和稳定性 [36] 。
5. 结论
本研究以隐–荧光Hf引物扩增5851 bp的隐–荧光HfDNA,Oligo和IDT为适合计算器。Hf引物能够扩增HfDNA,HfDNA-T5exo外切产物经激光共聚焦显微镜定性检测Fam荧光素,并用酶标仪定量检测荧光值,探讨了T5exo切割HfDNA的酶促动力学参数Km为0.1 nM,弥补了文献KD不足。结果表明HfDNA可以代替放射性标记DNA、非特异性荧光染料,为DNA酶学性质研究提供了新材料。
基金项目
国家自然科学基金面上项目(31771915)。
文章引用
刘自厚,刘亮伟. 隐–荧光DNA扩增
Amplification of Hidden Fluorescent DNA[J]. 微生物前沿, 2023, 12(01): 45-54. https://doi.org/10.12677/AMB.2023.121006
参考文献
- 1. Sayers, J.R. and Eckstein, F. (1990) Properties of Overexpressed Phage T5 D15 Exonuclease. Similarities with Escherichia coli DNA polymerase I 5’-3’ Exonuclease. The Journal of Biological Chemistry, 265, 18311-18317. https://doi.org/10.1016/S0021-9258(17)44753-3
- 2. Ma, X., Hong, Y., Han, W., et al. (2011) Single-Stranded DNA Binding Activity of XPBI, but Not XPBII, from Sulfolobus tokodaii Causes Double-Stranded DNA Melting. Extremophiles, 15, 67-76. https://doi.org/10.1007/s00792-010-0338-z
- 3. Joseph, J.W. and Kolodner, R. (1983) Exonuclease VIII of Escherichia coli. II. Mechanism of Action. The Journal of Biological Chemistry, 258, 10418-10424. https://doi.org/10.1016/S0021-9258(17)44473-5
- 4. Zhang, J., Xing, X., Herr, A.B. and Bell, C.E. (2009) Crystal Structure of E. coli RecE Protein Reveals a Toroidal Tetramer for Processing Double-Stranded DNA Breaks. Structure, 17, 690-702. https://doi.org/10.1016/j.str.2009.03.008
- 5. Richardson, C.C. (1966) The 5’-Terminal Nucleotides of T7 Bacteriophage Deoxyribonucleic Acid. Journal of Molecular Biology, 15, 49-61. https://doi.org/10.1016/S0022-2836(66)80208-5
- 6. Richardson, C.C., Inman, R.B. and Kornberg, A. (1964) Enzymatic Synthesis of Deoxyribonucleic Acid: XVIII. The Repair of Partially Single-Stranded DNA Templates by DNA Polymerase. Journal of Molecular Biology, 9, 46-69. https://doi.org/10.1016/S0022-2836(64)80090-5
- 7. Tolun, G. and Myers, R.S. (2003) A Real-Time DNase Assay (ReDA) Based on PicoGreen Fluorescence. Nucleic Acids Research, 31, e111. https://doi.org/10.1093/nar/gng111
- 8. Druml, B., Kaltenbrunner, M., Hochegger, R. and Cichna-Markl, M. (2016) A Novel Reference Real-Time PCR Assay for the Relative Quantification of (Game) Meat Species in Raw and Heat-Processed Food. Food Control, 70, 392-400. https://doi.org/10.1016/j.foodcont.2016.05.055
- 9. Carr, A.C. and Moore, S.D. (2012) Robust Quantification of Polymerase Chain Reactions Using Global Fitting. PLOS ONE, 7, e37640. https://doi.org/10.1371/journal.pone.0037640
- 10. Kubista, M., Andrade, J.M., Bengtsson, M., et al. (2006) The Real-Time Polymerase Chain Reaction. Molecular Aspects of Medicine, 27, 95-125. https://doi.org/10.1016/j.mam.2005.12.007
- 11. 马纪, 黄国霞, 姚运乾, 等. 光谱法结合凝胶电泳和荧光显微镜研究萘与DNA的相互作用及其应用[J]. 分析科学学报, 2022, 38(5): 582-590.
- 12. Wang, Z., Ouyang, H., Tesauro, C., et al. (2018) Real-Time Analysis of Cleavage and Religation Activity of Human Topoisomerase 1 Based on Ternary Fluorescence Resonance Energy Transfer DNA Substrate. Archives of Biochemistry and Biophysics, 643, 1-6. https://doi.org/10.1016/j.abb.2018.02.006
- 13. Kristoffersen, E.L., Jorgensen, L.A., Franch, O., et al. (2015) Real-Time Investigation of Human Topoisomerase I Reaction Kinetics Using an Optical Sensor: A Fast Method for Drug Screening and Determination of Active Enzyme Concentrations. Nanoscale, 7, 9825-9834. https://doi.org/10.1039/C5NR01474C
- 14. 柳苏月, 田晶晶, 朱龙佼, 等. 核酸外切酶Ⅲ辅助的DNA铽离子配合物时间分辨荧光检测黄曲霉毒素B1 [J]. 分析化学, 2021, 49(8): 1327-1334.
- 15. Marcussen, L.B., Jepsen, M.L., Kristoffersen, E.L., et al. (2013) DNA-Based Sensor for Real-Time Measurement of the Enzymatic Activity of Human Topoisomerase I. Sensors, 13, 4017-4028. https://doi.org/10.3390/s130404017
- 16. Zhao, B., Tong, Z.X., Zhao, G.J., et al. (2014) Effects of 2’-O-Methyl Nucleotide on Ligation Capability of T4 DNA Ligase. Acta Biochimica et Biophysica Sinica, 46, 727-737. https://doi.org/10.1093/abbs/gmu058
- 17. Li, X., Jin, J., Wang, M., et al. (2022) Abortive Ligation Intermediate Blocks Seamless Repair of Double-Stranded Breaks. International Journal of Biological Macromolecules, 209, 1498-1503. https://doi.org/10.1016/j.ijbiomac.2022.04.098
- 18. Saiki, R., Gelfand, D., Stoffel, S., et al. (1988) Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase. Science, 239, 487-491. https://doi.org/10.1126/science.2448875
- 19. Yang, A., Cheng, J., Liu, M., Shangguan, Y. and Liu, L. (2018) Sandwich Fusion of CBM9_2 to Enhance Xylanase Thermostability and Activity. International Journal of Biological Macromolecules, 117, 586-591. https://doi.org/10.1016/j.ijbiomac.2018.05.199
- 20. 黄亚威, 杨昂, 上官云杰, 等. 双退火温度PCR扩增DNA [J]. 微生物学报, 2017, 57(8): 1262-1269.
- 21. 邵玉强, 金家铖, 李雪刚, 刘亮伟. 大量-高纯-高浓度DNA的制备[J]. 微生物前沿, 2022, 11(2): 67-74.
- 22. 上官云杰, 梁亚萍, 杨昂, 等. 同源引物的非等量PCR [J]. 河南科学, 2018, 36(3): 326-333.
- 23. Jameson, E.E., Roof, R.A., Whorton, M.R., et al. (2005) Real-Time Detection of Basal and Stimulated G Protein GTPase Activity Using Fluorescent GTP Analogues. Journal of Biological Chemistry, 280, 7712-7719. https://doi.org/10.1074/jbc.M413810200
- 24. van Oijen, A.M., Glainey, P.C., Crampton, D.J., et al. (2003) Single-Molecule Kinetics of λ Exonuclease Reveal Base Dependence and Dynamic Disorder. Science, 301, 1235-1238. https://doi.org/10.1126/science.1084387
- 25. Bajar, B.T., Wang, E.S., Zhang, S., Lin, M.Z. and Chu, J. (2016) A Guide to Fluorescent Protein FRET Pairs. Sensors, 16, Article No. 1488. https://doi.org/10.3390/s16091488
- 26. Zhang, R., Kwok, R.T.K., Tang, B. and Liu, B. (2015) Hybridization Induced Fluorescence Turn-on of of AIEgen-Oli- gonucleotide Conjugates for Specific DNA Detection. RSC Advances, 5, 28332-28337. https://doi.org/10.1039/C5RA00322A
- 27. Cheng, Y., Stakenborg, T., Dorpe, P.V., et al. (2011) Fluorescence near Gold Nanoparticles for DNA Sensing. Analytical Chemistry, 83, 1307-1314https://doi.org/10.1021/ac102463c
- 28. Dubertret, B., Calame, M. and Libchaber, A.J. (2001) Single-Mismatch Detection Using Gold-Quenched Fluorescent Oligonucleotides. Nature Biotechnology, 19, 365-370. https://doi.org/10.1038/86762
- 29. Liu, F., Yang, Y., Wan, X., et al. (2022) Space-Confinment-Enhanced Fluorescence Detection of DNA on Hydrogel Particles Array. ACS Nano, 16, 6266-6273. https://doi.org/10.1021/acsnano.2c00157
- 30. 郑园霞, 张毅, 李雪刚, 刘亮伟. 荧光Cy5DNA扩增及酶切产物分离-检测[J]. 微生物前沿, 2022, 11(4): 241-250.
- 31. Spiliotis, M. (2012) Inverse Fusion PCR Cloning. PLOS ONE, 7, e35407. https://doi.org/10.1371/journal.pone.0035407
- 32. 韩来闯, 马闪闪, 刘亚娟, 等. 构建重组质粒的二步PCR方法[J]. 河南科学, 2015, 33(8): 1321-1325.
- 33. 刘猛, 刘亚娟, 徐文选, 等. 长片断引物反向 PCR 方法构建重复序列的重组质粒[J]. 河南科学, 2016, 34(4): 501- 505.
- 34. Breslauer, K.J., Frank, R., Blöcker, H. and Marky, L.A. (1986) Predicting DNA Duplex Stability from the Base Sequence. Proceedings of the National Academy of Sciences of the United States of America, 83, 3746-3750. https://doi.org/10.1073/pnas.83.11.3746
- 35. Allawi, H.T. and Santa, L.J. (1997) Thermodynamics and NMR of Internal G∙T Mismatches in DNA. Biochemistry, 36, 10581-10594. https://doi.org/10.1021/bi962590c
- 36. Takagi, M., Nishioka, M., Kakihara, H., et al. (1997) Characterization of DNA Polymerase from Pyrococcus sp. Strain KOD1 and Its Application to PCR. Applied and Environmental Microbiology, 63, 4504-4510. https://doi.org/10.1128/aem.63.11.4504-4510.1997
NOTES
*通讯作者。