Material Sciences
Vol.
09
No.
02
(
2019
), Article ID:
28905
,
6
pages
10.12677/MS.2019.92021
Molecular Dynamics Simulation for the Micro-Structure of MgO-Al2O3-SiO2 Glass-Ceramics
Yonghao Zhang, Jinlong Zhang, Shengwei Li, Kaixuan Cui, Jianjian Wang
School of Materials Science and Engineering, Shandong Jianzhu University, Jinan Shandong
Received: Jan. 31st, 2019; accepted: Feb. 12th, 2019; published: Feb. 19th, 2019

ABSTRACT
In this paper, MgO-Al2O3-SiO2 glass-ceramics were calculated by using a molecular dynamics method in this paper, and the microstructure characteristics of the system and the influence of SiO2 content on its network structure were obtained. It is shown by the calculation results that: In the MAS system at high temperature, the mean square displacement (MSD) of Mg2+ is the largest, followed by Al3+, followed by O2+ and the MSD of Si4+ is the smallest. The binding force between Si and O in MAS glass-ceramics is the strongest, followed by the force between Al and O, and the binding force between Mg and O is the weakest. The number of tetrahedrons is from more to less, followed by [SiO4] and [AlO4]. With the increase of SiO2 content, the number of [SiO4] tetrahedrons gradually increased, and the number of [AlO4] tetrahedrons increased at first and then decreased, and finally reached a state of equilibrium.
Keywords:MgO-Al2O3-SiO2 Glass-Ceramics, Molecular Dynamics Simulation, Micro-Structure, SiO2
MgO-Al2O3-SiO2系微晶玻璃微观结构的分子动力学模拟
张永豪,张金龙,李省伟,崔凯旋,王健健
山东建筑大学,材料科学与工程学院,山东 济南
收稿日期:2019年1月31日;录用日期:2019年2月12日;发布日期:2019年2月19日

摘 要
本文对MgO-Al2O3-SiO2 (MAS)微晶玻璃的高温体系的微观结构进行了分子动力学模拟,得出该体系微观体系结构特点以及SiO2含量对其网络结构的影响。计算结果显示:在MAS高温体系中,Mg2+的均方位移最大,其次是Al3+,然后是O2+,Si4+的均方位移最小。MAS系微晶玻璃中Si和O之间的作用力最强,Al和O之间的作用力次之,Mg和O之间的结合力最弱。四面体数量由多到少依次是[SiO4]、[AlO4],随着SiO2含量增多[SiO4]数量逐渐增加,[AlO4]数量随SiO2含量的增加呈现先增加然后减少最终趋向平稳的现象。
关键词 :MgO-Al2O3-SiO2系微晶玻璃,分子动力学模拟,微观结构,SiO2

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
微晶玻璃(玻璃陶瓷)多年来一直受到研究人员的广泛关注,其中MgO-Al2O3-SiO2 (MAS)系具有良好绝缘性和抗热震性、高机械强度、低介电常数等优异特性,可广泛应用于雷达天线罩、集成电路基片、硬盘基板和红外辐射材料等高新材料 [1] [2] [3] 。在微晶玻璃的发展中,人们越来越发现对微晶玻璃的高温微观结构研究的重要性,同时现在的分析测试手段不能完全了解微晶玻璃的高温微观结构。
在计算机的发展和多势体 [4] [5] [6] 研究深入的现在,更多的科研工作者开始利用分子动力学模拟来解决实验中难以解决的问题 [7] [8] [9] 。本文将通过Materials Studio对镁铝硅系微晶玻璃的高温融化状态进行分子动力学模拟研究,计算体系的高温微观结构特点及不同含量的SiO2对其微观体系结构的影响。
2. 模拟过程及参数设置
在计算过程中建立模型的首要原则是保证体系的电中性,同时需要保持体系中较高的硅含量,这样才可以形成致密的网络结构。在综合考虑计算机的计算能力和能代表MAS系微晶玻璃的粒子数量情况下,本次模拟确定了系统中粒子量保持在1300左右 [10] ,表1展示了每组中粒子的具体数量。

Table 1. Composition of seven different initial models of MAS system
表1. 七组体系初始模拟的具体数据
本次计算的降温机制如图1所示,降温过程为先让计算模型在6000 K下运行20 ps,得到无序和充分熔融的体系,然后用40 ps的时间降温至2000 K,在2000 K弛豫20 ps;然后经过20 ps时间冷却到1800 K,最后在1800 K温度下弛豫20 ps,保证体系处于稳定的高温熔体状态。
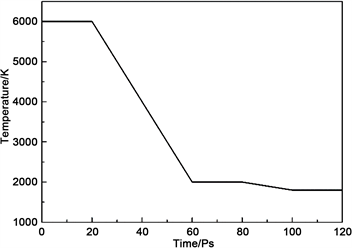
Figure 1. Temperature change in calculation
图1. 计算中的温度变化图
3. 结果和分析
3.1. 均方位移分析
图2是不同组各原子的均方位移(MSD)变化图。均方位移大小反映了体系中粒子的活跃程度,均方位移量越大,表明粒子在体系中越活跃,其他粒子对该粒子束缚力越小。反之,粒子在体系中位移量越小,表明粒子在体系中的活跃性差,其他粒子对该粒子的束缚能力强。从图中能够得出各组分中不同原子的均方位移量,它们的迁移速率关系均为Mg2+ > Al3+ > O2− > Si4+,这满足微晶玻璃内部粒子的运动特征。
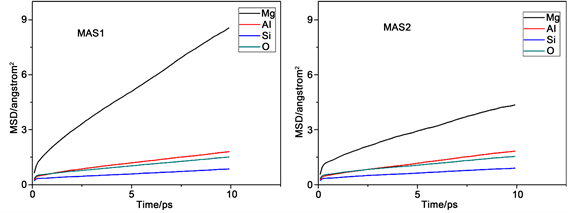
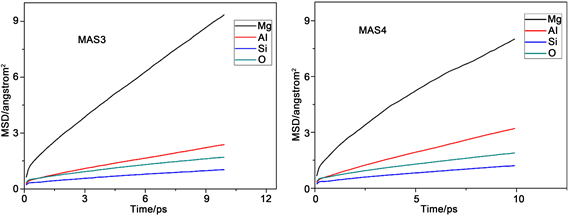
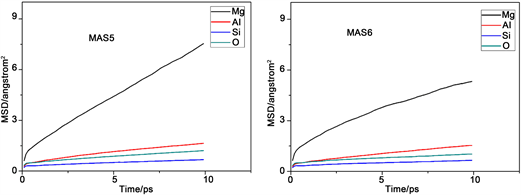
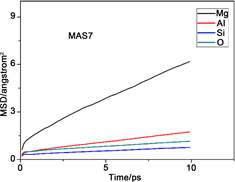
Figure 2. Mean square displacement (MSD) of various particles in the different component
图2. 不同组各粒子的均方位移(MSD)图
出现这种现象的原因是硅离子作为网络形成子参与了网络结构的构建,处于网络中间。镁离子处于游离态不参与网络结构组成,而铝离子作为网络中间体只有在特定条件下才会参与到结构中,有一部分不存在于网络结构之中,氧离子有部分作为桥氧处于网络内部,另外有部分作为非桥氧在网络的边缘活跃性高,所以氧离子的均方位移量比网络形成子硅离子大,但是由于体系中三氧的笼蔽效应导致了体系中氧离子的整体均方位移降低,所以铝离子的均方位移量比氧离子大。
3.2. 四面体分析
由图3可以得出,高温体系中[SiO4]的数量比[AlO4]的数量多。当SiO2含量增多时,该体系中[SiO4]数量逐渐增多,这表明[SiO4]内部作用力最大,受其他元素的影响小,Si和O具有比Al和O更强的结合能力。而[AlO4]数量随SiO2含量的增加呈现先增加然后减少最终趋向平稳的现象,并且在SiO2含量达到50.6%时,在镁铝硅玻璃陶瓷网络里[AlO4]数量达到最大值161个,出现这种现象的原因是铝离子是以网络中间体形式处于玻璃陶瓷网络结构中,只有在特定的情况下铝离子才可以进入网络结构中成为网络中间体。随着SiO2含量继续不断增加,网络结构变的越来越完整,此时[AlO4]的数量几乎不再变化。
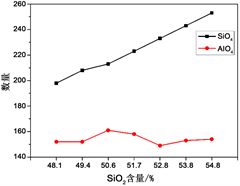
Figure 3. Change in the number of tetrahedrons of each component
图3. 各组四面体数量示意图
3.3. 径向分布函数分析
第四组分中,Mg、Al、Si三种粒子数之比为2:4:5,这是堇青石Mg2Al4Si5O18中的原子比,因此是一种较稳定的配方,所以以此组进行分析MAS系微晶玻璃微观熔融结构特征。
由图4可以看出Si-O、Al-O、Mg-O对应的第一峰峰形尖锐程度明显递减,表明Si和O两者的相互作用力最大,组成的团簇结构最牢固,Al和O的相互作用力次之,Mg和O之间作用力最小。并且同种原子之间没有第一尖锐峰,证明同种原子之间没有团聚现象。
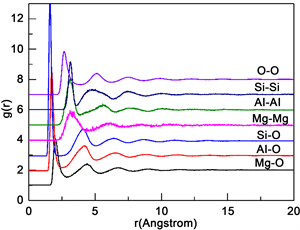
Figure 4. Radial distribution function (RDF) among various particles in the fourth group
图4. 第四组各粒子间的径向函数分布图
4. 结论
本文利用Materials Studio软件对MgO-Al2O3-SiO2微晶玻璃的高温体系的微观结构进行了分子动力学模拟,计算分析了该体系微观结构特点和SiO2含量对其网络结构的影响,得出以下结论:
1) 不同离子在MAS微晶玻璃熔融网络结构中均方位移大小关系为Mg2+ > Al3+ > O2− > Si4+。
2) 在MAS微晶玻璃高温体系中,[SiO4]的数量最多,其次是[AlO4]的数量,随着SiO2含量增多[SiO4]数量逐渐增加,[AlO4]数量随SiO2含量的增加呈现先增加然后减少最终趋向平稳的现象。
3) MAS系微晶玻璃中Si和O之间的作用力最强,Al和O之间的作用力次之,Mg和O之间的结合力最弱。
文章引用
张永豪,张金龙,李省伟,崔凯旋,王健健. MgO-Al2O3-SiO2系微晶玻璃微观结构的分子动力学模拟
Molecular Dynamics Simulation for the Micro-Structure of MgO-Al2O3-SiO2 Glass-Ceramics[J]. 材料科学, 2019, 09(02): 164-169. https://doi.org/10.12677/MS.2019.92021
参考文献
- 1. 郑孝英, 李毅恒, 冯康露, 何奥希, 陈菓. 堇青石基透波材料的抗热震性能分析[J]. 工业加热, 2016, 45(6): 17-19.
- 2. 张谦, 何涌. 粉煤灰堇青石玻璃陶瓷的抗热震性能[J]. 中国陶瓷, 2008, 44(5): 38-41.
- 3. 赵莹, 陆雷, 张朱军. MgO-Al2O3-SiO2系微晶玻璃的研究进展[J]. 新技术新工艺, 2006(9): 67-70.
- 4. Alfonso, P., Gianluca, M., Cristina, M.M., Cormack, A.N. and Ulderico, S. (2006) A New Self-Consistent Empirical Interatomic Potential Model for Oxides, Silicates, and Silica-Based Glasses. Journal of Physical Chemistry B, 110, 11780-11795. https://doi.org/10.1021/jp0611018
- 5. Okuno, M. and Kawamura, K. (1995) Molecular Dynamics Calculations for Mg3Al2Si3O12 (Pyrope) and Ca3Al2Si3O12 (Grossular) Glass Structures. Journal of Non-Crystalline Solids, 191, 249-259. https://doi.org/10.1016/0022-3093(95)00320-7
- 6. 丁元法, 张跃, 张凡伟, 张大海, 李仲平. 石英玻璃高温分子动力学模拟中的势函数[J]. 物理化学学报, 2008, 24(5): 788-792.
- 7. 罗旋, 费维栋, 李超, 姚忠凯. 材料科学中的分子动力学模拟研究进展[J]. 材料科学与工艺, 1996(1): 124-128.
- 8. 赵素, 李金富, 周尧和. 分子动力学模拟及其在材料科学中的应用[J]. 材料导报, 2007, 21(4): 5-8.
- 9. 王健健, 刘立强, 胡文广, 郝建林, 崔凯旋, 孙明杰. 分子动力学探究Fe~(3+)对CaO-Al_2O_3-SiO_2系微晶玻璃微观结构的影响[J]. 中国陶瓷, 2017(4): 34-37.
- 10. 李荣娟, 孙民华. 熔融ZnCl_2玻璃转变动力学减慢的分子动力学模拟[J]. 哈尔滨师范大学自然科学学报, 2009, 25(2): 62-64.