Open Journal of Natural Science
Vol.
09
No.
02
(
2021
), Article ID:
41249
,
9
pages
10.12677/OJNS.2021.92030
基于共同前缀位置的哺乳动物mtDNA序列 系统发育分析
陆超,王代
辽宁师范大学,辽宁 大连
收稿日期:2021年2月23日;录用日期:2021年3月22日;发布日期:2021年3月29日

摘要
本文基于共同前缀标识符,通过环形前缀树法构建DNA序列的环形前缀树,得到能够表征序列信息的前缀集,去除生物序列长度对序列间距离计算的影响的同时并考虑序列对的共同前缀位置等因素,创建了一种新的非比对方法——基于共同前缀的位置差法,选取含有80条哺乳动物的mtDNA序列的数据集作为测试对象,运用邻接(Neighbor-Joining)法构建系统发育树,快速且准确地得到符合生物学分类的结果。
关键词
哺乳动物,DNA序列,前缀集,系统发育分析
Phylogenetic Analysis of Mammalian mtDNA Sequences Based on Common Prefix Positions
Chao Lu, Dai Wang
Liaoning Normal University, Dalian Liaoning
Received: Feb. 23rd, 2021; accepted: Mar. 22nd, 2021; published: Mar. 29th, 2021

ABSTRACT
This paper is based on the common prefix identifier. A circular prefix tree of DNA sequences is constructed by the circular prefix tree method to obtain a prefix set that can characterize sequence information, which removed the influence of biological sequence length on the calculation of the distance between sequences, and considered the common prefix position of sequence pairs. And other factors, created a new non-comparison method—the position difference method based on the common prefix. The data set containing 80 mammalian mtDNA sequences was selected as the test object, and the phylogenetic tree was constructed using the Neighbor-Joining method, which could fast and accurately get the results that meet the biological classification.
Keywords:Mammal, DNA Sequence, PrefixSet, Phylogenetic Analysis

Copyright © 2021 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
20世纪50年代以来,随着分子生物学和分子遗传学的兴起,生物学家根据各种生物间在分子水平上的进化关系,建立分子进化的系统发育树(Phylogenetic Tree),估测物种间的亲缘关系,直观地阐明生物间的进化历程。
系统发育树重建传统上取决于多个或成对的序列比对。但是,使用基于对对的方法分析大型数据集时会遇到各种限制。高等真核生物的全基因组比对可能超过计算内存 [1]。因为存在基因组重排和重复的组合等因素使得不能进行整个基因组的比对,并且过多的序列信息增加了计算生物学中基因组比较的计算量和时间要求。因此,必须在初始步骤中确定正在研究的基因组的可比同源片段,并且迫切需要更快的序列分析算法。为此,提出了非比对方法克服基于比对方法的局限性 [2] [3] [4]。序列比较的非比对方法可以定义为在算法应用的任何步骤中量化序列相似性/相异性的任何方法,非比对方法不使用或产生比对(残基—残基对应的分配)。它们算法复杂度低且可以抵抗改组和重组等比对方法不能处理的事件。
非比对方法可大致分为两组:基于定义长度子序列频率的方法(基于词的方法)和评估全长序列之间信息内容的方法(基于信息理论的方法)。还有一些方法不能分类在任何一组中,包括那些基于匹配词长度的方法,混沌游戏表示,迭代映射,以及DNA序列的图形表示,它以定量的方式捕获基本组成的实质和序列的分布。本文是通过一种新的非比对方法——基于共同前缀的位置差算法(CPPA算法:Common Prefix Position-difference Algorithm)构建80中胎盘类哺乳动物的系统发育树,能够快速而准确地得到聚类结果,并且优于分布向量法 [5] 的结果。
2. 材料与方法
2.1. 材料
2020年10月22日从NCBI的Genebank数据库中下载的80条有胎盘类(Placentalia)哺乳动物的线粒体完整基因组,基因组的平均长度为1.65 × 103碱基对。因为它是在大多数多细胞生物中从母亲那里继承而来的,不是高度保守,而且突变率不是很高,所以它对于研究基于线粒体基因组的进化关系非常有效。本文将Bo等人(2011) [5] 中80个物种中含有非ACGT的序列进行了同属或同科替换,保证本方法的有效性,替换后的数据集详细信息见附录。
2.1.1. 前缀标识符
在有限字母 上指定的长度为m符号序列 ,其中 是序列中的第i个符号,并且被称为位于第i个位置。 表示从P的位置i开始到j的位置的字符子串。与 上的字符串S相关联的前缀树是t叉树 , 是 的最小长度唯一子串,其最左边的符号位于S的位置j。 在S中仅发生一次,而 在S中至少发生两次。则称 为与位置j相关的前缀标识符 [6]。
本文的CPPA算法的前缀树模型是以Weiner Peter的 [6] 前缀标识符理论为基础提出的,并通过将张欣 [7] 的相异度量模型中线性前缀树算法改进得到的。
2.1.2. 环形生物序列前缀集
哺乳动物的线粒体基因组是长度约为1.65 × 103碱基对的双链DNA环 [8]。但是从NCBI (National Center for Biotechnology Information Search database)网站中获取的序列是线性的,因此将序列首尾进行拼接,还原为环形序列,再基于前缀标识符,将线性前缀树算法 [7] 改进为环形前缀树算法构建前缀树,提取前缀集。
对任一序列 ,将序列S足够长的前m个碱基拼接在S末端,构建新序列 ,对 , 唯一前缀 ,s.t.当 时,满足 ,且至少存在一个 ,满足 。由 构成的集合 称为序列S的前缀集。
2.1.3. 序列间基于共同前缀的位置法
DNA序列 的长度分别为 。首先应用环形前缀树法构建两条序列的前缀树,得到前缀集 。再求出两条序列共同前缀集 的元素在 中的标准化位置,去除序列长度对计算序列间距离的影响。第t个共同前缀在 中的标准化位置定义为:
(1)
为第t个共同前缀在序列 中的实际位置。
假设 共有m个共同前缀,所有共同前缀在 中标准化位置构成如下位置向量: 。
两条序列的共同前缀的位置做差,得到位置差向量。考虑到哺乳动物的线粒体基因组是环形的,第t个共同前缀都的位置在序列中的位置如图1所示。
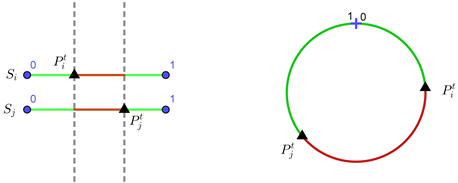
Figure 1. The relative position of the common prefix in the sequence pair
图1. 共同前缀在序列对中的相对位置
的第t个共同前缀的位置一同放在一个长度为1的环形DNA中,则距离差会出现两种结果红色弧线段的长度或绿色弧线段的长度,因为共同前缀位置之间的距离越小,则两序列越相似,因此取两种位置差中较小的数值作为所求的位置差。因此,两条序列 的位置差向量 定义为:
,其中 (2)
本文采用基于共同前缀的位置差算法来计算进化距离。因为序列间共同前缀越多,序列越相似,所以将序列对 间的相似性距离定义为:
(3)
获得了用于系统发育分析的所有遗传序列之间的距离构成的距离矩阵后,使用Matlab2013a通过邻接法(NJ)绘制系统发育树 [9]。
3. 结果
3.1. 系统发育分析
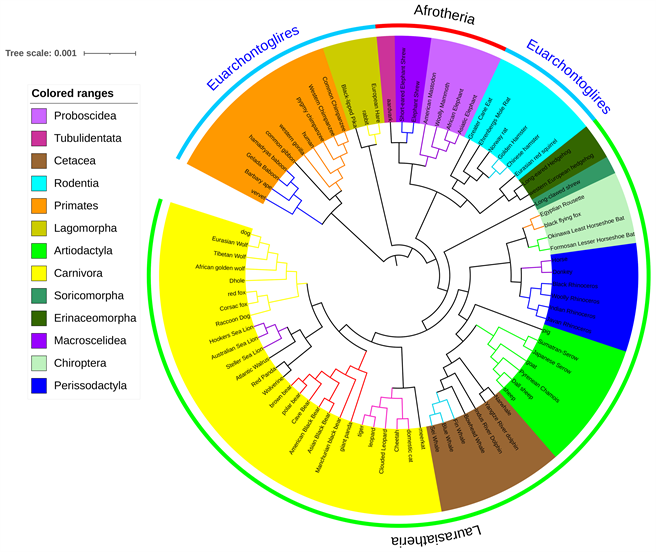
Figure 2. The Phylogenetic tree of 80 mammals by CPPA
图2. 由CPPA法得到的80种哺乳动物的系统发育树
80条哺乳动物线粒体全基因组mtDNA序列数据集中,全部是有胎盘类,其中非洲兽总目(Afrotheria) 7条序列,北方兽类(Boreoeutheria) 73条序列。北方兽类包含灵长总目(Euarchontoglires) 19条序列,劳亚兽总目(Laurasiatheria) 54条序列 [10] [11] [12]。
从整体上看,图2中CPPA法得到的系统发育树中,非洲兽总目正确聚类,灵长总目未聚为一个分支,劳亚兽总目的序列来自6个目,5个目的52条序列聚在一起;其它目的2条序列与灵长总目的啮齿目聚到一起。
数据集中,劳亚兽总目的序列具体是:食肉目(carnivora) 27条序列、鲸偶蹄目14条序列、奇蹄目(Perissodactyla) 6条序列、翼手目(Chiroptera) 4条序列、鼩形目(Soricomorpha) 1条序列和猬形目(Erinaceomorpha) 2条序列,6个目都分别聚成一个分支。食肉目的犬形亚目(Caniformia)与猫型亚目(Feliformia)分别聚为一个分支 [13],鲸偶蹄目中的3 条序列鲸目(cetacea)与偶蹄目(Artiodactyla)的6条反刍亚目(Ruminantia)和1条猪次目(Suina)正确聚类,并与交错在一起,支持鲸偶蹄目的分类 [14]。
灵长总目的序列来自3个目,灵长目(Primates)、兔形目(Lagomorpha)和啮齿目(Rodentia)。灵长总目虽然没有形成一个分支,灵长目和兔形目各自形成自己的分支,且聚类正确 [15]。啮齿目的6条序列来自3个亚目,鼠形亚目(Myomorpha) 4条序列,松鼠亚目(Sciuromorpha) 3条序列和豪猪亚目(Hystricomorpha) 1条序列 [16]。每个亚目各自形成单独的分支。
在科的层级上,含2条及以上的科共16个,均正确聚类。
3.2. CPPA法与分布向量法系统发育树比较
Bo Zhao和Rong L. He等人的分布向量法构建的系统发育树 [5],仅考虑了生物之间的聚类关系,因此本文在聚类的角度上进行比较。
一、目的层级上,分布向量法的聚类结果中27条食肉目序列被分为两部分,且猫型亚目与犬形亚目均未聚类正确,14条鲸偶蹄目序列被分为鲸目和偶蹄目两部分,不支持鲸偶蹄目的分类,CPPA法的食肉目与鲸偶蹄目均聚类正确,6条啮齿目序列的聚类CPPA法略差于分布向量法。
二、科的层级上,分布向量法的聚类结果中4条犀牛科(Rhinocerotidae)序列、3条海狮科(Otariidae)序列、7条犬科(Canidae)、3条须鲸科(Balaenopteridae)序列、5条人科(Hominidae)序列、4条猕猴科(Cercopithecidae)序列和2条兔科(Leporidae)序列7个科未聚类正确,CPPA法均聚类正确。
以上结果说明由80条哺乳动物线粒体全基因组mtDNA序列数据集构建的系统发育树中,CPPA法的聚类结果远好于分布向量法的聚类结果。
4. 结论
本文基于共同前缀标识符提出的CPPA法,首先通过环形前缀树算法获得每条序列的前缀集,挖掘到能表征序列的定量代表——共同前缀在序列对中所处的标准化位置,去除了序列长度对序列间距离的影响,并充分考虑了共同序列个数在计算序列间相似性距离的重要性,应用Matlab运行算法速度快,且聚类结果准确,说明CPPA法是一种好的非比对方法。通过CPPA法得到的系统发育树中物种间的进化关系还不够好,可以考虑影响序列间距离的其它因素进行改进。
致谢
感谢Bo等人为本研究提供了80条哺乳动物线粒体序列的NCBI检索号。同时,也要对本文中引用文章的作者表示衷心的感谢。
文章引用
陆 超,王 代. 基于共同前缀位置的哺乳动物mtDNA序列系统发育分析
Phylogenetic Analysis of Mammalian mtDNA Sequences Based on Common Prefix Positions[J]. 自然科学, 2021, 09(02): 272-280. https://doi.org/10.12677/OJNS.2021.92030
参考文献
- 1. Saw, A.K., Raj, G., Das, M., et al. (2019) Alignment-Free Method for DNA Sequence Clustering Using Fuzzy Integral Similarity. Scientific Reports, 9, Article No. 3753. https://doi.org/10.1038/s41598-019-40452-6
- 2. Vinga, S. and Almeida, J. (2003) Alignment-Free Sequence Comparison—A Review. Bioinforma, 19, 513-523. https://doi.org/10.1093/bioinformatics/btg005
- 3. Zielezinski, A., Vinga, S., Almeida, J. and Karlowski, W.M. (2017) Align-ment-Free Sequence Comparison: Benefits, Applications, and Tools. Genome Biology, 18, Article No. 186. https://doi.org/10.1186/s13059-017-1319-7
- 4. Bernard, G., Chan, C.X., Chan, Y.-B., et al. (2017) Alignment-Free Inference of Hierarchical and Reticulate Phylogenomic Relationships. Briefings in Bioinformatics, 20, 426-435.
- 5. Zhao, B., He, R.L. and Yau, S.S.-T. (2011) A New Distribution Vector and Its Application in Genome Clustering. Molecular Phylogenetics and Evolution, 59, 438-443. https://doi.org/10.1016/j.ympev.2011.02.020
- 6. Weiner, P. (1973) Linear Pattern Matching Algorithms. Proceedings of the 14th Annual IEEE Symposium on Switching and Automata Theory, USA, 15-17 October 1973, 1-11. https://doi.org/10.1109/SWAT.1973.13
- 7. 张欣. 基于后缀树的DNA序列进化树构建研究[D]: [硕士学位论文]. 大连: 辽宁师范学, 2019。
- 8. Anderson, S., De Bruijn, M.H.L., Coulson, A.R., et al. (1982) Complete Sequence of Bovine Mitochondrial DNA Conserved Features of the Mammalian Mitochondrial Genome. Journal of Molecular Biology, 156, 683-717. https://doi.org/10.1016/0022-2836(82)90137-1.
- 9. Saitou, N. and Nei, M. (1987) The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Molecular Biology and Evolution, 4, 406-425.
- 10. Murphy, W.J., Eizirik, E., Johnson, W.E., et al. (2001) Molecular Phylogenetics and the Origins of Placental Mammals. Nature, 409, 614-618. https://doi.org/10.1038/35054550
- 11. Kjer, K.M. and Honeycutt, R.L. (2007) Site Specific Rates of Mitochondrial Genomes and the Phylogeny of Eutheria. BioMed Central Evolutionary Biology, 7, Article No. 8. https://doi.org/10.1186/1471-2148-7-8
- 12. Xie, X.H., Yu, Z.G., Han, G.S., Yang, W.F. and Anh, V. (2015) Whole-Proteome Based Phylogenetic Tree Construction with Inter-Amino-Acid Distances and the Conditional Geometric Distribution Profiles. Molecu-lar Phylogenetics and Evolution, 89, 37-45. https://doi.org/10.1016/j.ympev.2015.04.008
- 13. Agnarsson, I., Kuntner, M. and May-Collado, L.J. (2010) Dogs, Cats, and Kin: A Molecular Species-Level Phylogeny of Carnivora. Molecular Phylogenetics and Evolution, 54, 726-745. https://doi.org/10.1016/j.ympev.2009.10.033
- 14. Zurano, J.P., Magalhães, F.M., Asato, A.E., et al. (2019) Cetartiodactyla: Updating a Time-Calibrated Molecular Phylogeny. Molecular Phylogenetics and Evolution, 133, 256-262. https://doi.org/10.1016/j.ympev.2018.12.015
- 15. Dos Reis, M., Gunnell, G.F., Barba-Montoya, J., et al. (2018) Using Phylo-genomic Data to Explore the Effects of Relaxed Clocks and Calibration Strategies on Divergence Time Estimation: Primates as a Test Case. Systematic Biology, 67, 594-615. https://doi.org/10.1093/sysbio/syy001
- 16. Steppan, S.J. and Schenk, J.J. (2017) Muroid Rodent Phylogenetics: 900-Species Tree Reveals Increasing Diversification Rates. PLoS ONE, 12, e0183070. https://doi.org/10.1371/journal.pone.0183070
附录

Table 1. Description of mitochondrial genome of 80 mammalians
表1. 80条哺乳动物线粒体序列