Hans Journal of Medicinal Chemistry
Vol.
11
No.
03
(
2023
), Article ID:
70797
,
8
pages
10.12677/HJMCe.2023.113024
靶向CDK6抗肿瘤小分子抑制剂的虚拟筛选
李树香1,王敏寿1*,赵静峰2,王雁声1,徐高鑫1
1滇西应用技术大学乡村振兴学院,云南 大理
2云南大学化学科学与工程学院,云南 昆明
收稿日期:2023年7月14日;录用日期:2023年7月24日;发布日期:2023年8月17日

摘要
目的:细胞周期蛋白依赖性激酶6 (Cyclin-dependent kinases 6, CDK6)过表达导致的细胞周期异常是导致癌细胞过度增殖的原因,阻断CDK6表达可抑制肿瘤存活和生长。因此,CDK6抑制剂被认为是一种很有前途的抗癌药物。运用不同的虚拟筛选方法,从Specs数据库中筛选具有CDK6抑制活性的成分。方法:在本研究中,设计了针对特定靶标CDK6的虚拟筛选流程。为了发现结构新颖的CDK6抑制剂,通过类药性评估、基于蛋白质–配体复合物的药效团搜索、分子对接技术等多轮虚拟筛选策略,从Specs数据库中寻找命中分子。结果:通过类药性初筛,共获得75,671个小分子。类药性化合物进行最佳药效团匹配得到630个小分子用于下一步的分子对接研究。最终保留了对接打分高于已知抑制剂且结合模式与已知抑制剂相似的2个小分子为最佳命中分子。结论:本研究为新一代CDK6抑制剂的开发提供研究方向。
关键词
虚拟筛选,药效团,分子对接,CDK6激酶

Virtual Screening of Antitumor Small Molecule Inhibitors Targeting CDK6
Shuxiang Li1, Minshou Wang1*, Jingfeng Zhao2, Yansheng Wang1, Gaoxin Xu1
1Department of Rural Revitalization, West Yunnan University of Applied Sciences, Dali Yunnan
2School of Chemical Science and Technology, Yunnan University, Kunming Yunnan
Received: Jul. 14th, 2023; accepted: Jul. 24th, 2023; published: Aug. 17th, 2023

ABSTRACT
Objective: The abnormal cell cycle caused by Cyclin-dependent kinases 6 (CDK6) overexpression is responsible for excessive proliferation of cancer cells, and blocking CDK6 expression can inhibit tumor survival and growth. Therefore, CDK6 inhibitors are considered to be promising anticancer drugs. To screen the components withCDK6 inhibitory activity from the Specs database using different virtual screening methods. Methods: In this study, a virtual screening process for CDK6 was designed. In order to discover CDK6 inhibitors with novel structures, multiple rounds of virtual screening strategies, such as drug-like evaluation, complex-based pharmacophore searching based on protein-ligand structure and molecular docking technology, were used to explore the hit molecules from Specs database. Result: A total of 75671 small molecules were obtained through the drug-like preliminary screening. Drug-like compounds were used to match the best receptor-ligand pharmacophore hypothesis, as a result, 630 common compounds were identified for further molecular docking. Finally, the two small molecules whose docking scores were higher than known inhibitors and binding patterns were similar to known inhibitors were retained as the best hit molecules. Conclusion: This study provides research direction for the development of a new generation of CDK6 inhibitors.
Keywords:Virtualscreening, Pharmacophore, Molecular Docking, CDK6 Kinase

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
全球范围内癌症发病率持续上升,2020年全球确诊癌症患者数达1929万人,近1000万人死于癌症 [1] 。有大量研究表明CDK4/6在多种肿瘤细胞中被过度激活或扩增,包括乳腺癌、黑色素瘤、鳞状细胞癌、骨肉瘤、皮肤癌、膀胱癌、肺癌和胃癌和白血病等 [2] [3] 。CDK4/CDK6是细胞周期的关键调节因子,过表达的CDK4/6通过与细胞周期蛋白D形成复合物,磷酸化视网膜母细胞瘤基因,触发细胞周期从DNA合成前期(G1期)进入到DNA复制期(S1期),促进肿瘤的发生 [4] 。据报道,CDK6具有不同于CDK4的独特功能,并且参与调控细胞代谢和细胞分化、转录以及DNA修复。在CDK6高表达的肿瘤细胞系内使用CDK6抑制剂可阻止肿瘤细胞从G1期进展到S期并诱导肿瘤细胞凋亡,进而达到抑制肿瘤细胞增殖的目的 [5] [6] 。抑制CDK6是一种很有前景的癌症治疗策略,CDK4/6双重抑制剂palbociclib、ribociclib和abemaciclib已被FDA批准用于治疗晚期或转移性乳腺癌患者 [7] [8] ,dalpiciclib已被我国药品监督管理局上市用于转移性乳腺癌患者,trilaciclib是FDA批准的全球首个在化疗期间预防性给药以保护骨髓和免疫系统功能 [9] (图1)。接二连三的CDK6抑制剂药物的上市,证明其作为新的癌症治疗靶点是成功的。然而,CDK6抑制剂的结构缺乏多样性,经典抑制剂都含有一个N-苯基嘧啶-2-胺结构,不能克服临床应用中的副作用 [10] [11] 。为了丰富结构多样性,探索更多潜在的CDK6抑制剂,我们采用基于结构的药物设计策略来设计新型CDK6选择性抑制剂。由于CDK6抑制剂在许多癌症中的潜在应用,有效的CDK6抑制剂的发现和设计近年来引起了研究人员广泛的关注,高效的药物设计对于未来癌症的治疗非常有意义。
2. 材料与方法
2.1. 数据集准备
Specs 数据库(http://www.specs.net)总共包含212,770个小分子化合物,用做筛选的目标数据库,使用Discovery Studio 2016 (DS)软件进行三维结构的准备和优化。为了摒除那些不适合成为药物的分子,利用里宾斯基和韦伯规则(Lipinski and Veber rules)对Specs数据库进行类药性初筛 [12] [13] 。根据分辨率,从PDB网站(https://www.rcsb.org/)下载CDK6的晶体结构5L2S [4] (分辨率:2.27Å),用来构建基于配体–受体复合物的药效团模型以及分子对接研究。下载的蛋白结构使用DS进行预处理,删除无关水分子,补全缺失的残基,加上氢原子并添加CHARM力场。
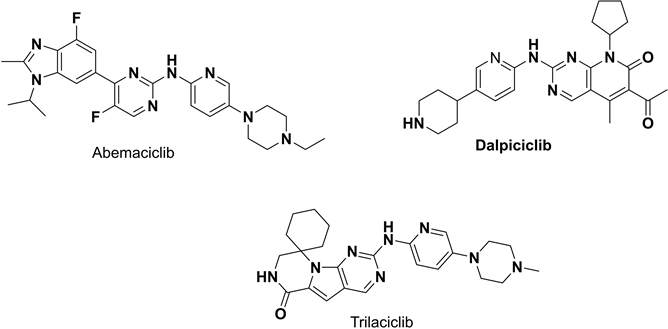
Figure 1. CDK4/6 inhibitors currently on the market
图1. 目前已上市的CDK4/6抑制剂
准备具有已知抑制剂和非抑制剂的测试集,用于后续分析。我们从PubChem网站 (https://pubchem.ncbi.nlm.nih.gov/)下载了200个已知的CDK6抑制剂,并从pubchem的3D结构数据库随机下载了25,000个小分子。下载的25,000个随机小分子通过DS中Find diverse structure得到结构多样性的4000个分子作为假定的非活性分子。以200已知的抑制剂和4000个假定非活性分子,组成一个验证数据库,用于评价药效团模型区分活性分子和非活性分子的能力。
2.2. 方法
2.2.1. 药效团模型构建
为了精简Specs数据库,使用DS软件构建基于复合物的药效团模型来快速筛选数据库。该药效团根据复合物的受体–配体相互作用信息,得到抑制剂中对活性贡献较大的基团及其空间分布特征并将此信息转化成药效团模型,以此为搜寻条件,得到符合药效团特征的优选化合物。该药效团使用DS软件中内置的Receptor-Ligand Pharmacophore Generation模块进行建模。建模选用的受体–配体复合物5L2S是人源的CDK6激酶和已上市药物Abemaciclib结合的晶体结构。设置测试集分子,包括活性分子与非活性分子;小分子以CHARMm力场的Best模式产生构象,其余参数采用默认设置。
2.2.2. 药效团模型的验证
用建立的测试集评价模型的敏感性(sensitivity, SE)和特异性(specificity, TN)。前者用于判断药效团的选择性,得分越高,选择性越好。后者用于验证模型区分活性分子和非活性分子的能力。模型计算得到的敏感性和特异性的值越高,说明其识别活性分子的能力越强。可从ROC (receiver operating characteristic)曲线对这两个量化指标进行综合评估。对于每个模型都具有相应的ROC曲线图,用曲线下面积值(area under the curve, AUC)代表最后的统计结果。AUC越大,模型识别能力越强 [14] 。该值应大于0.5,越大代表模型区分能力越强。
2.2.3. 虚拟筛选
在基于药效团的虚拟筛选中,使用打分结果评价小分子与模型的匹配程度。用DS中的LPM (pharmacophore mappingprotocol)模块以Best/Flexible Search选项,并将maximum omitted features设置为0。最佳药效团模型作为三维提问结构对Specs数据库进行筛选。根据化合物构象与药效团特征叠合程度,计算其拟合值(Fit values),其值越高,则代表小分子化合物与药效团模型匹配越好。
2.2.4. 分子对接及聚类分析
为获得更可靠和准确的虚拟筛选结果进行更准确的分子对接。使用DS软件中的分子对接程序CDOCKER进行半柔性对接。CDOCKER基于格点的分子对接方法,采用高温动力学产生配体的多构象,小分子结合构象采用模拟退火进行优化 [15] ,通过计算受体–配体的相互作用能及配体的张力来进行对接打分。在对接过程中考虑结合位点和小分子的柔性,可以产生高精度的对接结果。将小分子对接到CDK6的活性口袋中,基于晶体配体Abemaciclib (PDB ID:5L2S)定义活性位点,设置半径为10Å,其他参数设置为默认值。综合考虑对接打分及结合模式,选取对接打分高于已知抑制剂且与已知抑制剂结合模式相似的分子作为潜在的CDK6抑制剂。
3. 结果
3.1. 药效团的构建与验证
基于受体–配体复合物,提取关键的相互作用并转化为药效团元素,最终产生10个药效团模型(表1)。10个药效团的药效特征元素包括氢键受体(A)、氢键供体(D)、疏水(H)、正电离中心(P)。在该药效团构建过程中采用了两种验证方法,包括用表1中Selectivity Score的值来判断药效团的选择性,分值越高说明选择性越高,以及用已知活性化合物和非活性化合物建立的验证数据集来验证模型区分活性和非活性化合物的能力。通过运算,保留前10个Selectivity Score值最高的模型。

Table 1. The results of receptor-ligand pharmacophores
表1. 基于受体–配体药效团建模结果
由于Selectivity Score值并没有验证模型筛选的能力,我们建立验证集来对10个药效团模型的筛选能力进行分析,做出ROC曲线。模型2的AUC值最高为0.977,表明该模型具有最佳的识别活性分子和非活性分子的能力,因此选择模型2作为下一步筛选模型(图2(a))。模型2包含2个氢键受体,1个氢键供体,1个正电离(P)和1个疏水特征(图2(b))。
 图注:(a) 药效团模型2的ROC曲线图;(b) 模型2的药效团特征,绿色表示氢键受体,红色代表正电中心,蓝色表示疏水特征,紫色代表氢键给体,灰色为排除体积。
图注:(a) 药效团模型2的ROC曲线图;(b) 模型2的药效团特征,绿色表示氢键受体,红色代表正电中心,蓝色表示疏水特征,紫色代表氢键给体,灰色为排除体积。
Figure 2. The evaluation results of hypothesis 2
图2. 药效团模型2的评估结果
3.2. 虚拟筛选结果
根据类药规则初筛,得到75,671个小分子。经过验证的药效团模型2用于从75,671个小分子中搜索有效的CDK6抑制剂,可以匹配最佳药效团模型2的化合物有630个。
3.3. 分子对接研究
将虚拟筛选得到的化合物对接到CDK6的结合口袋中,根据对接结果,选取了打分值高于Abemaciclib的34个化合物进行进一步的结合模式研究。据文献报道,CDK6激酶和小分子抑制剂的相互作用主要是与氨基酸残基VAL101和ASP163形成氢键相互作用,与LEU152、ILE19等形成广泛的疏水相互作用 [4] 。进一步分析了化合物与CDK6激酶的相互作用,选择了2个化合物作为最终的虚拟筛选命中分子,其结构如图3所示。如图4所示,对接分析统计了所有体系中,与氢键和疏水相互作用有关的氨基酸残基,发现相互作用主要集中在残基LEU152、ILE19、VAL101,结果与文献报道的结果一致。
进一步分析了化合物与CDK6的相互作用,如图5所示。化合物AO-476/43222921与残基ILE19、VAL101和ASP104形成氢键相互作用,主要与ILE19和LEU152形成疏水相互作用。化合物AO-022/43452190与GLU18、VAL101、LYS147、ASP163等形成氢键相互作用,与ILE19、LEU152、VAL27等形成广泛的疏水相互作用。结果表明,2个分子的相互作用模式与Abemaciclib相似。这表明2种候选物可能是有效的CDK6抑制剂。
利用谷本系数TC (Tanimoto coefficient) [16] 评估2个分子骨架的新颖性。根据以前的研究,TC值高于0.85的结构可以被认为与已知的CDK6抑制剂相似。通过相似性分析,2个命中分子与已知的抑制剂相比,其TC系数分别为0.631858 (AO-022/43452190)和0.544228 (AO-476/43222921),均不大于0.7。这表明2个命中分子和已知的活性化合物结构明显不同,结构具有新颖性。
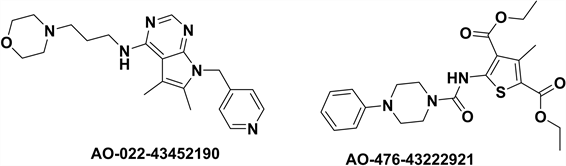
Figure 3. Chemical structures of hit molecules
图3. 命中分子化学结构
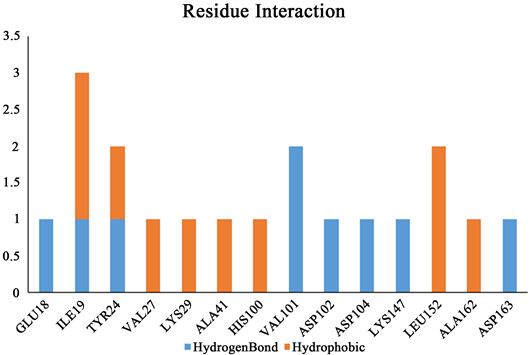
Figure 4. The analysis of critical residues in docking study
图4. 对接的关键残基分析
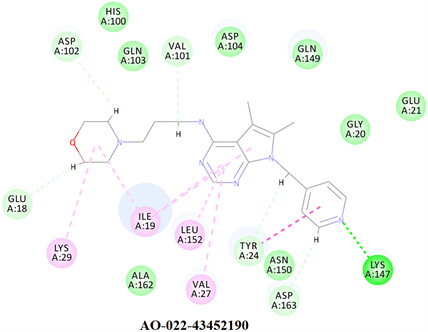
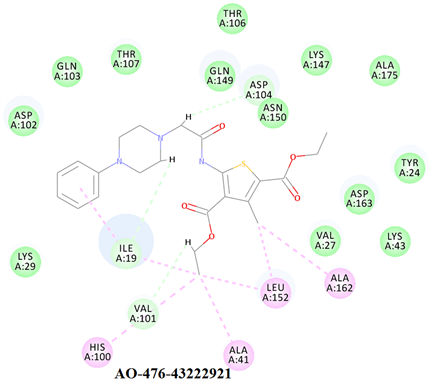
Figure 5. The putative binding modes of compounds with CDK6
图5. 化合物与CDK6的预测结合模式
4. 结论
计算机辅助药物设计从大量的有机化合物中有效地遴选出先导化合物,避免了盲目的活性筛选,从而降低了发现活性先导化合物的人力、时间和财力成本。通过药效团模型和分子对接研究相结合的方法,从化合物Specs数据库中鉴定出2种具有不同结构的新型CDK6抑制剂。分子对接结果表明,有2个化合物的结合模式与已知抑制剂相似,且分子对接打分高于已知的抑制剂。通过相似性计算,2个命中分子结构与已知抑制剂结构明显不同,最终获得了2个骨架新颖的CDK6激酶潜在的抑制剂。但上述化合物的激酶抑制活性尚需进行生物实验进一步验证。本研究可为CDK6激酶抑制剂的开发提供研究思路及新化合物骨架参考。
基金项目
云南省科技厅科技计划——基础研究专项青年基金(202201AU070092)资助。
文章引用
李树香,王敏寿,赵静峰,王雁声,徐高鑫. 靶向CDK6抗肿瘤小分子抑制剂的虚拟筛选
Virtual Screening of Antitumor Small Molecule Inhibitors Targeting CDK6[J]. 药物化学, 2023, 11(03): 196-203. https://doi.org/10.12677/HJMCe.2023.113024
参考文献
- 1. Ferlay, J., et al. (2021) Cancer Statistics for the Year 2020: An Overview. International Journal of Cancer, 149, 778-789. https://doi.org/10.1002/ijc.33588
- 2. Zhang, J., Yang, P.L. and Gray, N.S. (2009) Targeting Cancer with Small Molecule Kinase Inhibitors. Nature Reviews Cancer, 9, 28-39. https://doi.org/10.1038/nrc2559
- 3. Müller, S., Chaikuad, A., Gray, N.S. and Knapp, S. (2015) The Ins and Outs of Selective Kinase Inhibitor Development. Nature Chemical Biology, 11, 818-821. https://doi.org/10.1038/nchembio.1938
- 4. Chen, P., et al. (2016) Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Molecular Cancer Therapeutics, 15, 2273-2281. https://doi.org/10.1158/1535-7163.MCT-16-0300
- 5. O’Leary, B., Finn, R.S. and Turner, N.C. (2016) Treating Cancer with Selective CDK4/6 Inhibitors. Nature Reviews Clinical Oncology, 13, 417-430. https://doi.org/10.1038/nrclinonc.2016.26
- 6. Finn, R.S., et al. (2020) Biomarker Analyses of Response to Cy-clin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clinical Cancer Research, 26, 110-121. https://doi.org/10.1158/1078-0432.CCR-19-0751
- 7. Eggersmann, T.K., Degenhardt, T., Gluz, O., Wuerstlein, R. and Harbeck, N. (2019) CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs, 33, 125-135. https://doi.org/10.1007/s40259-019-00337-6
- 8. Zhu, L., et al. (2022) Novel Therapeutic Strategy for Melanoma Based on Albendazole and the CDK4/6 Inhibitor Palbociclib. Scientific Reports, 12, Article No. 5706. https://doi.org/10.1038/s41598-022-09592-0
- 9. Roskoski Jr., R. (2022) Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2022 Update. Pharmacological Research, 175, Article ID: 106037. https://doi.org/10.1016/j.phrs.2021.106037
- 10. Yuan, K., et al. (2021) Selective Inhibition of CDK4/6: A Safe and Effective Strategy for Developing Anticancer Drugs. Acta Pharmaceutica Sinica B, 11, 30-54. https://doi.org/10.1016/j.apsb.2020.05.001
- 11. Guarducci, C., et al. (2017) Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer and Potential Biomarkers of Response. Breast Care, 12, 304-308. https://doi.org/10.1159/000484167
- 12. Veber, D.F., et al. (2002) Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry, 45, 2615-2623. https://doi.org/10.1021/jm020017n
- 13. Lipinski, C.A., Lombardo, F., Dominy, B.W. and Feeney, P.J. (2001) Ex-perimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews, 46, 3-26. https://doi.org/10.1016/S0169-409X(00)00129-0
- 14. Lätti, S., Niinivehmas, S. and Pentikäinen, O.T. (2016) Rocker: Open Source, Easy-to-Use Tool for AUC and Enrichment Calculations and ROC Visualization. Journal of Cheminformatics, 8, 45. https://doi.org/10.1186/s13321-016-0158-y
- 15. 夏侯真如, 薛孟祺, 汪欣怡, 等. 基于分子对接及药效团模型的木姜子属木脂素类化合物5-LOX抑制剂筛选研究(I) [J]. 云南大学学报(自然科学版), 2022, 44(2): 369-379.
- 16. Hutter, M.C. (2022) Differential Multimolecule Fingerprint for Similarity Search-Making Use of Active and Inactive Compound Sets in Virtual Screening. Journal of Chemical Information and Modeling, 62, 2726-2736. https://doi.org/10.1021/acs.jcim.2c00242
NOTES
*通讯作者。