Applied Physics
Vol.05 No.02(2015), Article ID:14851,7
pages
10.12677/APP.2015.52002
Quasiparticle Band Structure and Optical Absorption Spectrum of Alkaline-Earth Oxide CaO
Bo Pan, Nengping Wang
Department of Microelectronic Science and Engineering, Science Faculty, Ningbo University, Ningbo Zhejiang
Email: panbo1988@126.com, wangnengping@nbu.edu.cn
Received: Feb. 1st, 2015; accepted: Feb. 13th, 2015; published: Feb. 16th, 2015
Copyright © 2015 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
This paper reports the quasiparticle band structure and the optical absorption spectrum of alkaline-earth metal oxide CaO, using many-body perturbation theory. The quasiparticle band structure is calculated within the GW approximation. Taking the electron-hole interaction into consideration, electron-hole pair states and optical excitations are obtained by solving the Bethe-Salpeter equation for the electron-hole two-particle Green function. The calculated band gap for CaO is 7.3 eV, which is in good agreement with the experimental results of 7.1 eV. The theoretical result of optical absorption spectrum for CaO is also in agreement with the experimental data. In particular, the calculated excitation energy for the lowest exciton peak in the optical absorption spectrum of CaO reproduces the corresponding experimental result very well.
Keywords:Quasiparticle Band Structure, Density-Functional Theory, Local Density Approximation, GW Corrections, Electron-Hole Interaction, Optical Absorption Spectra, Bethe-Salpeter Equation
碱土金属氧化物CaO的准粒子能带及光吸收谱
潘播,王能平
宁波大学理学院微电子科学与工程系,浙江 宁波
Email: panbo1988@126.com, wangnengping@nbu.edu.cn
收稿日期:2015年2月1日;录用日期:2015年2月13日;发布日期:2015年2月16日

摘 要
本文使用多体摄动理论计算了碱土金属氧化物CaO的准粒子能带结构和光吸收谱。我们运用GW近似来改进密度泛函理论的局域密度近似对电子交换关联的处理,并计算了CaO准粒子能带。对于电子-空穴激发态,求解二粒子格林函数的Bethe-Salpeter方程可计算电子-空穴激发态和光吸收谱。计算得出CaO的能隙是7.3 eV,与实验结果7.1 eV符合较好。CaO光吸收谱的理论结果与实验数据也一致。特别是在计算CaO激发能最低的激子峰时,计算结果很好地再现了实验结果。
关键词 :准粒子能带,密度泛函理论,局域密度近似,GW修正,电子-空穴相互作用,光吸收谱,Bethe-Salpeter方程

1. 引言
碱土金属氧化物代表了典型的离子晶体。他们是重要的材料,广泛应用于在微电子、催化、自旋电子、光电子等领域。为了这些应用,我们需要完全理解电子激发态。密度泛函理论(DFT)包括局域密度近似(LDA) [1] [2] 或广义梯度近似(GGA)已经被证明是一个非常有效的工具,成功计算了许多材料电子基态的属性。但是密度泛函理论严重低估了半导体和绝缘体的带隙[3] [4] 。
多体摄动理论(MBPT)是一种计算激发电子态的严密方法[5] -[9] 。激发态看作是相对于电子基态的准粒子激发,可用对应的格林函数来描述。我们通过求解单粒子格林函数的运动方程来确定电子态和空穴态,并求解出体系的能带结构。而通过求解二粒子格林函数的运动方程(即Bethe-Salpeter方程,BSE)可计算材料的电子-空穴激发态。基于DFT对基态的计算结果,准粒子激发(包括电子或空穴激发)可由Hedin的GW近似(GWA) [10] [11] 来精确描述。GWA对许多半导体和绝缘体的带隙计算结果与实验结果相符合,GW方法已成为预言准粒子能带的一种标准工具。因半导体和绝缘体的光激发受到电子-空穴关联效应的影响[5] ,Bethe-Salpeter方程就被用来求解这种激发态。在计算耦合电子-空穴激发态的光跃迁矩阵元后,就可容易得出光吸收谱。这种方法已经成功地应用于计算各种材料的光吸收谱[6] -[9] [13] -[22] 。
最近,一种自相互作用修正DFT(即SIC-DFT)方法被用来改善标准LDA的能带结构,这种方法部分地解决了LDA带隙问题[23] -[28] 。但是,对于LiF和一些其他绝缘体,SIC-DFT的计算结果明显地低估了其最高价带和最低导带的带宽。因为这些能带的色散关系会影响激子态的结构,带隙和能带色散对理解材料的光学性质也同样重要。论文[29] ,分别运用基于SIC-DFT能带结构来求解Bethe-Salpeter方程,以及多体摄动理论(即基于GWA能带结构来求解Bethe-Salpeter方程)两种方法,来计算LiF和其他一些绝缘体的光吸收谱。由于SIC-DFT对最低导带和最高价带带宽的计算结果明显小于GWA的相应计算结果,采用SIC-DFT电子能量和波函数作为BSE输入而得到的光吸收谱与实验数据相比,吸收峰向低能发生明显的移动;而用MBPT方法计算的结果与实验测量的光吸收谱很好地符合[28] [29] 。
论文[30] ,在线性光学的角度,用正交原子轨道线性叠加(OLCAO)基的LDA方法研究了碱土金属氧化物和其他材料的线性光学谱。论文[31] ,用全势能线性缀加平面波GGA的方法研究了碱土金属氧化物的光吸收谱。然而,这些计算碱土金属氧化物的光吸收谱[30] [31] 与实验结果[32] 并不符合(见下文)。这表明,他们[31] [32] 对CaO的计算忽略了一些重要的物理效应。为了给CaO光激发性质提供一个准确的理论结果,我们使用多体摄动理论计算了CaO的能带结构和光吸收谱。特别是,我们将研究CaO中电子-空穴相互作用的效应,激子态的两粒子波函数在实空间的特征。
本文结构如下:在下节中将简要描述理论方法,在第三节中,我们将给出准粒子能带结构和光吸收谱的计算结果,并与实验数据相比较。第四节,我们将对论文进行总结。
2. 理论计算方法
2.1. 准粒子能带计算方法
我们运用密度泛函理论[2] 的标准局域密度近似来计算体系的基态性质,其中所采用的赝势是非局域的模守恒赝势[33] [34] 。在构造赝势时,对于O,将其2s,2p态作为价态;而对于Ca,将其4s态作为价态。本文中,我们用s,p,d,s*四个高斯轨道来构建Kohn-Sham波函数的基矢[35] 。在这些高斯轨道中,Ca的衰减常数是0.17,0.7,1.5,O的衰减常数是0.21,1.15,3.1。在涉及GW自能运算和电子-空穴相互作用计算时,我们采用相同的基失。我们通过对Kohn-Sham方程的自洽运算获得了LDA的能带结构,波函数和总能量。
DFT-LDA的交换关联能不能正确地描述固体中的电子关联,因此,我们采用多体摄动理论来研究准粒子的激发,交换关联用一个能量相关的非局域自能算符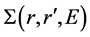 来描述。这样,我们得到如下准粒子方程[36] :
来描述。这样,我们得到如下准粒子方程[36] :
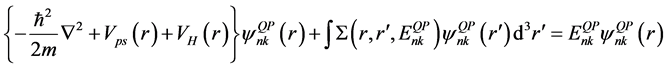 (1)
(1)
这里,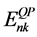 和
和 分别表示准粒子能量和准粒子波函数。自能算符由GWA [10] [11] [37] 来计算
分别表示准粒子能量和准粒子波函数。自能算符由GWA [10] [11] [37] 来计算
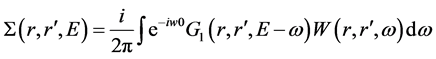 (2)
(2)
其中,G1和W分别为单粒子格林函数和屏蔽库伦作用。
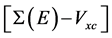 表示GWA对LDA能带结构的修正。在实际GWA计算中,单粒子格林函数G1近似地由DFT-LDA的单粒子能量和波函数来计算。我们运用广义等离激元-极点近似(generalized plasmon-pole approximation) [36] [38] 来描述屏蔽库伦作用W对频率的依赖关系;而W的零频项则用无规相近似(RPA)进行计算。
表示GWA对LDA能带结构的修正。在实际GWA计算中,单粒子格林函数G1近似地由DFT-LDA的单粒子能量和波函数来计算。我们运用广义等离激元-极点近似(generalized plasmon-pole approximation) [36] [38] 来描述屏蔽库伦作用W对频率的依赖关系;而W的零频项则用无规相近似(RPA)进行计算。
2.2. 光吸收谱计算方法
由于光激发(即电子-空穴激发)不能用单粒子激发来描述,这是由于激发的电子和空穴之间存在相互作用。为了描述光激发,我们需要引进二粒子激发态。利用Tamm-Dancoff [39] ,电子-空穴激发态可表示为
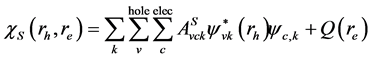 (3)
(3)
表式(3)中S标志电子-空穴激发态,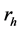 和
和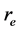 分别表示空穴和电子的坐标。Q为电子-空穴态的总动量,相当于吸收光子的动量。由于光子的波长比晶体的晶格常数要大的多,光子的总动量Q在光吸收中非常接近于零。但是,光子动量Q的方向在光吸收谱计算中具重要性。在表式(3)中,
分别表示空穴和电子的坐标。Q为电子-空穴态的总动量,相当于吸收光子的动量。由于光子的波长比晶体的晶格常数要大的多,光子的总动量Q在光吸收中非常接近于零。但是,光子动量Q的方向在光吸收谱计算中具重要性。在表式(3)中,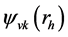 和
和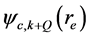 分别为电子和空穴的波函数。
分别为电子和空穴的波函数。
使用二粒子格林函数的运动方程,电子空穴对振幅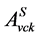 及相应的激发能
及相应的激发能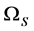 可由如下Bethe-Salpeter方程[12] [40] 决定:
可由如下Bethe-Salpeter方程[12] [40] 决定:
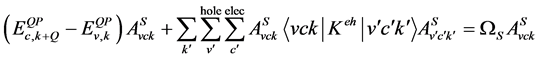 (4)
(4)
在此, 为电子-空穴相互作用核 [12] [40] 。
为电子-空穴相互作用核 [12] [40] 。
在解完BSE后,我们由如下宏观横向介电函数的虚部来计算光吸收谱:
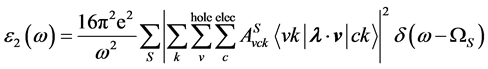 (5)
(5)
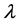 是入射光的极化方向,
是入射光的极化方向, 是单粒子速度算符,其中
是单粒子速度算符,其中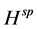 是单粒子哈密顿量。如果忽略电子-空穴相互作用,所得到的激发是独立的空穴带和电子带之间竖直跃迁,相应的光吸收谱由下式给出:
是单粒子哈密顿量。如果忽略电子-空穴相互作用,所得到的激发是独立的空穴带和电子带之间竖直跃迁,相应的光吸收谱由下式给出:
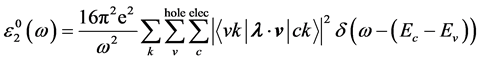 (6)
(6)
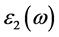 减去
减去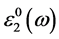 的值表示电子-空穴相互作用对光吸收谱的影响。
的值表示电子-空穴相互作用对光吸收谱的影响。
3. 计算结果
CaO是一种典型的岩盐结构绝缘体,它的实验晶体结构常数是4.811埃[41] 。为了与实验结果相比较,本文在计算CaO的准粒子能带和光吸收谱时,都将采用实验的晶体结构常数。
3.1. 能带计算
通过自洽求解Kohn-Sham方程,沿着布里渊区的高对称点,我们得到了CaO的LDA能带结构,并用虚线表示在图1中。通过GW修正,我们用方程(1)计算了CaO的准粒子能带,用实线表示在图1中。GWA的价带共有四个带组成,−17.5 eV到−16.8 eV之间归属于O的2s带,三条O的2p带在−3.3 eV至0 eV之间(最大值在Γ点)。最低的导带在7.3 eV至11.0 eV之间(最小值在X点),其主要包括了Ca的4s轨道(也包含了小部分的O 3s轨道的贡献)。O的2p带和Ca的4s带的LDA带宽分别为2.8 eV和3.5 eV;通过准粒子修正,分别得到GWA带宽3.3 eV和3.7 eV。能隙也从LDA的3.7 eV增加到GWA的7.3 eV。能隙的实验结果是7.1 eV [42] ,我们的GWA能隙结果与实验能隙吻合得很好。
3.2. 光吸收谱
利用上一节所述的计算光吸收谱的方法,我们计算了CaO的光吸收谱。在计算中,我们采用了4条价带和最低的8条导带,在布里渊区取了500个k点;这样,电子-空穴相互作用矩阵大小为4 × 8 × 500 = 16000。求解方程(4)后,介电函数的虚部可以由方程(5)得到。求得的光吸收谱用实线展示在图2中。通过增加k点和引入更多导带的再计算,我们发现上述计算已收敛。也就是说,取500个k点,引入4条价带和8条导带已足够描述低激发能(即小于12eV)的CaO光吸收谱。我们理论计算得到的第一个吸收峰在7.4 eV。图1中GWA计算得到的最小直接带隙是7.6 eV (位于 点),这个值比第一个激发峰的能量大了0.2 eV。因此,图2中第一个吸收峰对应的激子态具有结合能0.2 eV。为了比较,我们把实验结果[32] 也放在图2中,实验结果的第一个吸收峰在7.3 eV。很明显,光吸收谱的主峰,无论从激发能还是从强度来看都与实验值结果符合得很好。在低于12 eV的低激发能区时,我们计算出的光吸收谱的形状与实验结果符合得也不错。
点),这个值比第一个激发峰的能量大了0.2 eV。因此,图2中第一个吸收峰对应的激子态具有结合能0.2 eV。为了比较,我们把实验结果[32] 也放在图2中,实验结果的第一个吸收峰在7.3 eV。很明显,光吸收谱的主峰,无论从激发能还是从强度来看都与实验值结果符合得很好。在低于12 eV的低激发能区时,我们计算出的光吸收谱的形状与实验结果符合得也不错。
为了展示忽略电子-空穴作用的光吸收谱结果,本文也把这种计算结果用绿色的粗点曲线表示在图2中。显然,电子-空穴作用严重影响整个吸收谱的结果,如果电子-空穴相互作用被忽视,那么第一个吸收峰将上移约2.5 eV,且强度小很多。而考虑了电子-空穴作用以后,理论吸收谱与实验吸收谱符合度就大大提高了。图2中也展示了论文[30] [31] 中CaO不考虑电子-空穴相互作用的理论计算结果,与他们[30] [31]
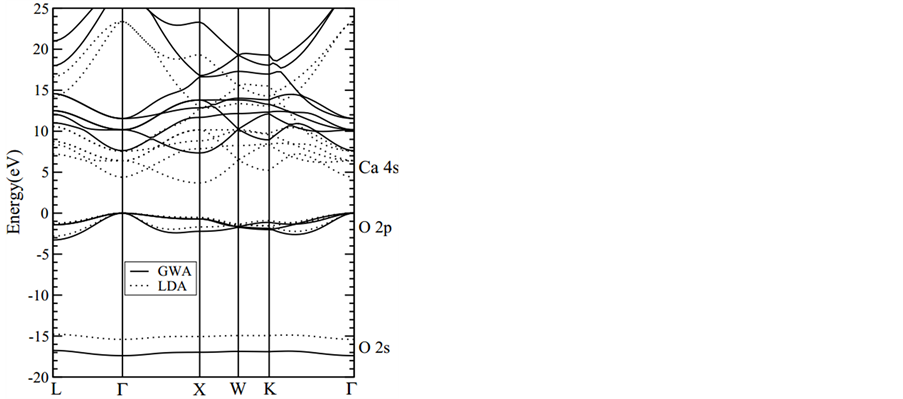
Figure 1. Bulk band structure of CaO, as calculated within LDA (dashed curves) and within GWA (solid curves)
图1. CaO的能带结构图,虚线是LDA的计算结果,实线是GWA的计算结果
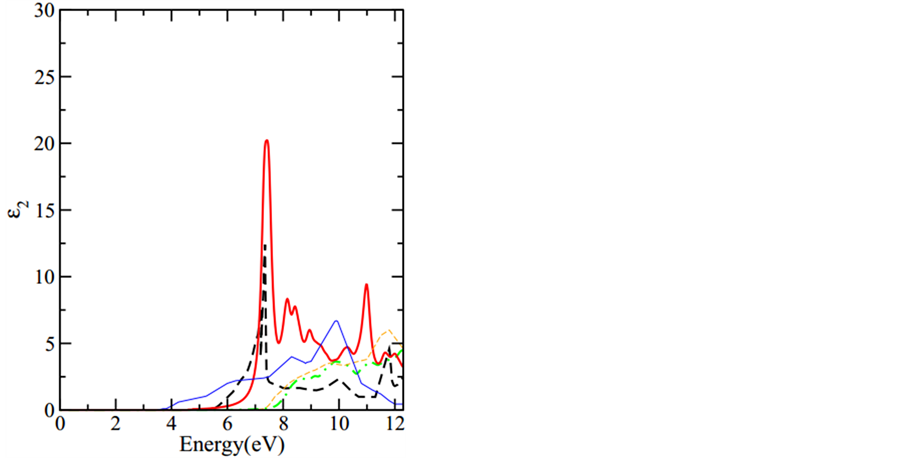
Figure 2. The calculated optical absorption spectrum of bulk CaO is compared with experimental measurements [32] (thick black dashed curve). Thick red solid curve: optical spectrum (calculated with the electron-hole interaction included) obtained by this work. Thick green dash-dotted curve, thin blue solid curve, and thin orange dashed curve: independent-particle spectrum (i.e., without electron-hole interaction) obtained by this work and those taken from Ref [30] and [31] , respectively.
图2. CaO理论光吸收谱和实验吸收谱图,黑色的粗虚线是实验值[32] ,红色的粗实线是本文(考虑电子-空穴相互作用)的计算结果。绿色的粗点曲线,蓝色的细实线,橙色的细短划线分别是本文(不考虑电子-空穴相互作用)和论文[30] [31] 的计算结果
相比我们的计算结果与实验的一致程度要好的多。
3.3. 最低激子态的波函数
现在,我们考查激子态波函数的空间特性。使用系数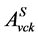 ,电子-空穴激发态的波函数
,电子-空穴激发态的波函数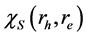 也可以从公式(3)中求得。
也可以从公式(3)中求得。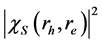 在电子和空穴之间给出了一个实空间的关联。我们计算了最低激子态(激发能为7.4 eV)的概率密度
在电子和空穴之间给出了一个实空间的关联。我们计算了最低激子态(激发能为7.4 eV)的概率密度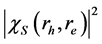 。图3展示的是在(001)平面电子相对空穴的实空间分布,其中空穴
。图3展示的是在(001)平面电子相对空穴的实空间分布,其中空穴

Figure 3. Real-space distribution 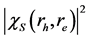 of the electron
of the electron  with respect to the hole
with respect to the hole  for the lowest-energy exciton of CaO (at 7.4 eV), in the (001) plane. The hole
for the lowest-energy exciton of CaO (at 7.4 eV), in the (001) plane. The hole 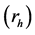 is fixed at the central O atom
is fixed at the central O atom
图3. CaO最低激子态(激发能为7.4 eV)的电子相对空穴在(001)平面的实空间的密度分布图。空穴固定在中间O原子上
固定在中间O原子。电子态主要来自于附近O原子的3s和很少部分的Ca原子4s轨道的贡献。
通过图3,我们可以在电子的空间分布上发现一些有意义的特性。把空穴固定在中间O原子上时,电子的概率密度在中间的O原子上达到最大值,在周围的O原子上也相当大。然而,在Ca原子上,密度就相对很低。既然在Ca原子上的概率密度低,那么此时的激子态就不是简单的Frenkel激子。此外,电子在相邻的各个O原子上的分布也不是各向同性的,而是偏向中间的O原子。
4. 结论
本文通过多体摄动理论,研究了碱土金属氧化物CaO块体的电子激发态。首先,我们用局域密度近似下的密度泛函理论来计算出体系的基态性质。然后用GW近似,计算了体系的准粒子能带结构。最后,我们考虑了电子-空穴相互作用,求解BSE方程,计算了CaO的光吸收谱。我们得出的GWA能隙与实验结果[42] 符合得好,光吸收谱中最低激子态所形成的第一个吸收峰也与实验结果[32] 符合得很好,当然整个光吸收谱的形状也与实验结果符合得不错。此外,我们展现了CaO最低激子态的电子空间分布图。最低激子态电子分布图中呈现了一个很明显的极化特性, 此特性归因于电子-空穴相互作用。
考虑电子-空穴相互作用极大地改善了理论光吸收谱与实验结果[32] 符合程度。比较忽略考虑电子-空穴相互作用的光吸收谱,我们发现,在计算碱土金属CaO的光吸收谱时,必须考虑电子-空穴相互作用。
基金项目
感谢中国国家自然科学基金项目(项目编号11074136)和浙江省自然科学基金项目(项目编号6100467)对本研究工作的资助。
文章引用
潘 播,王能平, (2015) 碱土金属氧化物CaO的准粒子能带及光吸收谱
Quasiparticle Band Structure and Optical Absorption Spectrum of Alkaline-Earth Oxide CaO. 应用物理,02,9-16. doi: 10.12677/APP.2015.52002
参考文献 (References)
- 1. Hohenberg, P. and Kohn, W. (1964) Inhomogeneous electron gas. Physical Review, 136, B864-B871.
- 2. Kohn, W. and Sham, L.J. (1965) Self-consistent equations including exchange and correlation effects. Physical Review, 140, A1133-A1138.
- 3. Ohno, K., Esfarjani, K. and Kawazoe, Y. (1999) Computational materials science. Springer, Ber-lin.
- 4. Onida, G., Reining, L. and Rubio, A. (2002) Electronic excitations: Density-functional versus many-body green’s- function approaches. Reviews of Modern Physics, 74, 601-659.
- 5. Hanke, W. and Sham. L.J. (1980) Many-particle effects in the optical spectrum of a semiconductor. Physical Review B, 21, 4656-4673.
- 6. Hybertsen, M.S. and Louie, S.G. (1985) First-principles theory of quasiparticles: Calculation of band gaps in semiconductors and insulators. Physical Review Letters, 55, 1418-1421.
- 7. Onida, G., Reining, L., Godby, R.W., Del Sole, R. and An-dreoni, W. (1995) Ab initio calculations of the quasiparticle and absorption spectra of clusters: The sodium tetramer. Physical Review Letters, 75, 818-821.
- 8. Benedict, L.X., Shirley, E.L. and Bohn, R.B. (1998) Optical absorption of insulators and the electron-hole interaction: An ab initio calculation. Physical Review Letters, 80, 4514-4517.
- 9. Rohlfing, M. and Louie, S.G. (1998) Excitonic effects and the optical absorption spectrum of hydro-genated Si clusters. Physical Review Letters, 80, 3320-3323.
- 10. Hedin, L. (1965) New method for calculating the one-particle Green’s function with application to the electron-gas problem. Physical Review, 139, A796-A823.
- 11. Hedin, L. and Lundqvist, S. (1969) Solid state physics: Advances in research and application. Aca-demic Press, New York.
- 12. Strinati, G. (1982) Dynamical shift and broadening of core excitons in semiconductors. Physical Review Letters, 49, 1519-1522.
- 13. Rohlfing, M. and Louie, S.G. (1999) Excitons and optical spectrum of the Si(111)-(2 × 1) surface. Physical Review Letters, 83, 856-859.
- 14. vander Horst, J.W., Bobbert, P.A., Michels, M.A.J., Brocks, G. and Kelly, P.J. (1999) Ab initio calculation of the electronic and optical excitations in polythiophene: Effects of intra- and interchain screening. Physical Review Letters, 83, 4413-4416.
- 15. Grossman, J.C., Rohlfing, M., Mitas, L., Louie, S.G. and Cohen, M.L. (2001) High accuracy many-body calculational approaches for excitations in molecules. Physical Review Letters, 86, 472-475.
- 16. Hahn, P.H., Schmidt, W.G. and Bechstedt, F. (2002) Bulk excitonic effects in surface optical spectra. Physical Review Letters, 88, 164021-164024.
- 17. Wang, N.P., Rohlfing, M., Krüger, P. and Pollmann, J. (2003) Quasiparticle band structure and optical spectrum of LiF(001). Physical Review B, 67, Article ID: 115111.
- 18. Wang, N.P., Rohlfing, M., Krüger, P. and Pollmann, J. (2004) Electronic excitations of CO adsorbed on MgO(001). Applied Physics A, 78, 213-221.
- 19. Wang, N.P., Rohlfing, M., Krüger, P. and Pollmann, J. (2004) Image states and excitons at insulator surfaces with negative electron affinity. Physical Review Letters, 91, Article ID: 216805.
- 20. Wang, N.P., Rohlfing, M., Krüger, P. and Pollmann, J. (2006) Electronic excitations of the H:Si(001)-(2 × 1) monohydride surface: First-principles calculations. Physical Review B, 74, Article ID: 155405.
- 21. Ma, Y., Rohlfing, M. and Gali, A. (2010) Excited states of the negatively charged nitrogen-vacancy color center in diamond. Physical Review B, 81, Article ID: 041204(R).
- 22. Rohlfing, M. (2012) Redshift of excitons in carbon nanotubes caused by the environment polarizability. Physical Review Letters, 108, Article ID: 087402.
- 23. Vogel, D., Krüger, P. and Pollmann, J. (1996) Self-interaction and relaxation-corrected pseudopotentials for II-VI semiconductors. Physical Review B, 54, 5495-5511.
- 24. Filippetti, A. and Fiorentini, V. (2009) A practical first-principles band-theory approach to the study of correlated materials: Self-interaction corrected local-density-functional theory. European Physical Journal B, 71, 139-183.
- 25. Erwin, S.C. and Lin, C.C. (1988) The self-interaction-corrected electronic band structure of six alkali fluoride and chloride crystals. Journal of Physics C, 21, 4285-4309.
- 26. Baumeier, B., Krüger, P. and Pollmann, J. (2007) Bulk and surface electronic structures of alkaline-earth metal oxides: Bound surface and image-potential states from first principles. Physical Review B, 76, Article ID: 205404.
- 27. Baumeier, B., Krüger, P., Pollmann, J. and Vajenine, G.V. (2008) Electronic structure of alkali-metal fluorides, oxides, and nitrides: Density-functional calculations including self-interaction corrections. Physical Review B, 78, Article ID: 125111.
- 28. Sommer, C., Krüger, P. and Pollmann, J. (2012) Optical spectra of alkali-metal fluorides. Physical Review B, 86, Article ID: 155212.
- 29. Sommer, C., Krüger, P. and Pollmann, J. (2012) Quasiparticle band structure of alkali-metal fluorides, oxides, and nitrides. Physical Review B, 85, Article ID: 165119.
- 30. Ching, W.Y., Gan, F. and Huang, M.Z. (1995) Band theory of linear and nonlinear susceptibilities of some binary ionic insulators. Physical Review B, 52, 1596-1611.
- 31. Dadsetani, M. and Beiranvand, R. (2009) Optical properties of alkaline-earth metal oxides from first principles. Solid State Sciences, 11, 2099-2105.
- 32. Kaneko, Y., Morimoto, K. and Koda, T. (1983) Optical properties of alkaline-earth chalcogenides. II. Vacuum ultraviolet reflection spectra in the synchrotron radiation region of 4-40 eV. Journal of the Physical Society of Japan, 52, 4385-4396.
- 33. Hamann, D.R. (1989) Generalized norm-conserving pseudopotentials. Physical Review B, 40, 2980-2987.
- 34. Bachelet, G.B., Hamann, D.R. and Schlüter, M. (1982) Pseudopotentials that work: From H to Pu. Physical Review B, 26, 4199-4228.
- 35. Rohlfing, M., Krüger, P. and Pollmann, J. (1995) Efficient scheme for GW quasiparticle band-structure calculations with applications to bulk Si and to the Si(001)-(2 × 1) surface. Physical Review B, 52, 1905-1917.
- 36. Rohlfing, M., Krüger, P. and Pollmann, J. (1993) Quasiparticle band-structure calculations for C, Si, Ge, GaAs, and SiC using Gaussian-orbital basis sets. Physical Review B, 48, 17791-17805.
- 37. Hybertsen, M.S. and Louie, S.G. (1988) Model dielectric matrices for quasiparticle self-energy calculations. Physical Review B, 37, 2733-2736.
- 38. Hybertsen, M.S. and Louie, S.G. (1986) Electron correlation in semiconductors and insulators: Band gaps and quasiparticle energies. Physical Review B, 34, 5390-5413.
- 39. Fetter, A. and Walecka, J.D. (1973) Quantum theory of many particle systems. McGraw-Hill, San Francisco.
- 40. Rohlfing, M. and Louie, S.G. (2000) Electron-hole excitations and optical spectra from first principles. Physical Review B, 62, 4927-4944.
- 41. Madelung, O. (2004) Semiconductors: Data handbook. 3rd Edition, Springer, Berlin.
- 42. Whited, R.C., Flaten, C.J. and Walker, W.C. (1973) Exciton thermoreflectance of MgO and CaO. Solid State Communications, 13, 1903-1905.