Applied Physics
Vol.06 No.02(2016), Article ID:17184,7
pages
10.12677/APP.2016.62003
First-Principles Studies of the Al, Ga, In-Doped ZnO Defect Formation Energy
Yuhang Qi, Li Niu*, Qi Guan, Huamei Xu, Huiqing Lu, Chunqiu You
Key Laboratory for Photonic and Electronic Bandgap Materials, Ministry of Education, Institute of Physics and Electronic Engineering, Harbin Normal University, Harbin Heilongjiang

Received: Mar. 4th, 2016; accepted: Mar. 20th, 2016; published: Mar. 23rd, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
The defect formation energy and the defect transition energy level as well as electronic energy band structure of IIIA (Al, Ga and In)-doped ZnO crystal were investigated by density functional calculations using local density approximation + Hubbard U (LDA + U) approach. We discussed the stability and ionization properties of doped ZnO crystal. Alternative doping in ZnO crystal introduces a shallow donor level so that be ionized easily. GaZn and Gai has a low formation energy and the crystal structure is relatively stable. The conduction band of the doped ZnO is slightly decreased, the Fermi level moves into the conduction band.
Keywords:First-Principles, LDA + U, Doped ZnO, Electronic Energy Band Structure, Defect Formation Energy

Al、Ga、In掺杂ZnO形成能的第一性原理研究
祁雨杭,牛丽*,关启,许华梅,卢会清,由春秋
哈尔滨师范大学物理与电子工程学院,光电帯隙省部共建教育部重点实验室,黑龙江 哈尔滨

收稿日期:2016年3月4日;录用日期:2016年3月20日;发布日期:2016年3月23日

摘 要
利用LDA + U方法计算ⅢA族元素Al、Ga、In掺杂ZnO晶体的能带结构、形成能和跃迁能级,讨论ⅢA族元素掺杂ZnO晶体结构的稳定性和电离性质。替代掺杂在ZnO晶体中形成一个浅施主能级,容易发生电离;GaZn和Gai的形成能相对较低,晶体结构相对稳定;掺杂后ZnO导带下移,费米能级穿过导带。
关键词 :第一性原理,LDA + U,掺杂ZnO,能带结构,形成能

1. 引言
透明导电氧化物(TCO)属于半导体材料,具有较大的禁带宽度、可见光谱透过、红外区域高反射率以及较低的电阻率等良好的光电特性,因而被广泛的应用于太阳能电池 [1] 、显示器 [2] 、特殊功能窗口涂层及其它光电器件领域。作为透明导电氧化物材料之一的氧化锌(ZnO)有着与氮化镓(GaN)相似的晶体结构和禁带宽度,其高达60 meV [3] 的激子束缚能是GaN的2.7倍,具备室温机子发光的特性,光电器件方面ZnO比GaN有更大的应用潜力。
纤锌矿ZnO是Ⅱ-Ⅵ族直接带隙半导体,室温下禁带宽度为3.37 eV。Al、Ga、In [4] 掺杂ZnO很容易获得n型半导体 [5] [6] ,作为浅施主能级提高了ZnO的电子导电性,提高了透明高能电子设备电子和光学性能 [7] 。实验表明ZnO掺杂ⅢA族元素可以增加光禁带宽度和电导率而且不会明显减少光传输的能力 [8] [9] 。
缺陷形成能是有关费米能级、化学势与价带顶的函数,表示各种晶格缺陷形成的难易程度、缺陷材料的稳定性等。通过比较缺陷形成能的函数图像,可以直观的看出:1) 一种物质掺杂的形成能随费米能级变化情况;2) 同种物质不同掺杂位置时缺陷形成能的大小;3) 不同物质相同掺杂位置时缺陷形成能的大小。
本文计算了Al、Ga、In三种原子在ZnO超晶格体系中替代Zn原子掺杂以及间隙掺杂的形成能,试图通过比较它们的形成能函数图像和跃迁能级来讨论Al、Ga、In三种原子掺杂ZnO晶体的稳定性和电离性。
2. 计算结构和方法
近来,人们使用多种方法计算了掺杂ZnO的形成能,如基于密度泛函理论的LDA、HSE、PBE [10] 等方法 [11] 。从理论计算结果来看,LDA方法计算得出的ZnO带隙只有0.80 eV,严重低估了半导体体系的带隙宽度。除此之外,计算结果强烈依赖于晶胞尺寸。为了可靠的预测掺杂ZnO体系的形成能,必须在一定程度上克服以上两个问题。本文使用VASP软件的LDA+U计算方法对Zn原子3d轨道电子进行+U处理,分别计算Al、Ga、In掺杂ZnO的总能以及In掺杂ZnO的能带结构,U = 4.7 eV。
2.1. 能带结构
选用2 × 2 × 2的32个原子的超胞来计算能带结构,特殊k点网格取5 × 5 × 3,截断能取550 eV。用一个In原子替代ZnO超胞中一个Zn原子,In掺杂ZnO晶胞的浓度为6.25%。
2.2. 总能计算
使用4 × 3 × 2的96个原子的ZnO超胞作为掺杂的超单胞,特殊k点取![]() ,优化结构后所有原子受力都小于0.01 eV/Å,价电子分别取为Zn原子3d4s、Al原子3s3p、Ga原子3d4s4p和In原子4d5s5p轨道电子。考虑掺杂原子替代晶胞中心一个Zn原子和间隙原子处于晶胞八面体中心位置两种结构,分别计算它们的总能。在32原子或72原子ZnO晶胞中掺杂原子,发现它与不同方向相邻超单胞中杂质的间距不同,不具有对称性,而且在结构优化时也不易收敛;而在96原子超晶格中(图1),晶胞基矢互相垂直,具有正交对称性,避免了32原子和72原子ZnO超胞的问题 [12] 。
,优化结构后所有原子受力都小于0.01 eV/Å,价电子分别取为Zn原子3d4s、Al原子3s3p、Ga原子3d4s4p和In原子4d5s5p轨道电子。考虑掺杂原子替代晶胞中心一个Zn原子和间隙原子处于晶胞八面体中心位置两种结构,分别计算它们的总能。在32原子或72原子ZnO晶胞中掺杂原子,发现它与不同方向相邻超单胞中杂质的间距不同,不具有对称性,而且在结构优化时也不易收敛;而在96原子超晶格中(图1),晶胞基矢互相垂直,具有正交对称性,避免了32原子和72原子ZnO超胞的问题 [12] 。
2.3. 缺陷形成能
缺陷形成能的定义式:
![]() (1)
(1)
式中![]() 为超单胞的总能;
为超单胞的总能;![]() 为掺杂超单胞的总能(α = Al、Ga或In)。
为掺杂超单胞的总能(α = Al、Ga或In)。![]() 为原子的化学势,
为原子的化学势,![]() 为掺杂晶体中减少(
为掺杂晶体中减少(![]() )或增加(
)或增加(![]() )的原子数,q是电子转移的数量。
)的原子数,q是电子转移的数量。![]() 是相对于超单胞能带结构价带顶(VBM)能量
是相对于超单胞能带结构价带顶(VBM)能量![]() 的费米能级。
的费米能级。
原子化学势 的大小取决于生长条件,分为富锌与富氧两个极端情况。在本文中我们认为ZnO、Al2O3、Ga2O3和In2O3的形成来自于金属Zn、Al、Ga、In和O2的直接化合,基于这个假设推导出化学势:
的大小取决于生长条件,分为富锌与富氧两个极端情况。在本文中我们认为ZnO、Al2O3、Ga2O3和In2O3的形成来自于金属Zn、Al、Ga、In和O2的直接化合,基于这个假设推导出化学势:
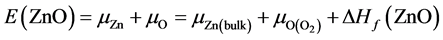 (2)
(2)
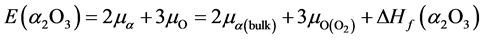 (3)
(3)
富锌
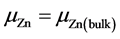 (4)
(4)
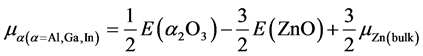 (5)
(5)
富氧
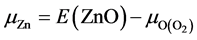 (6)
(6)
 (7)
(7)
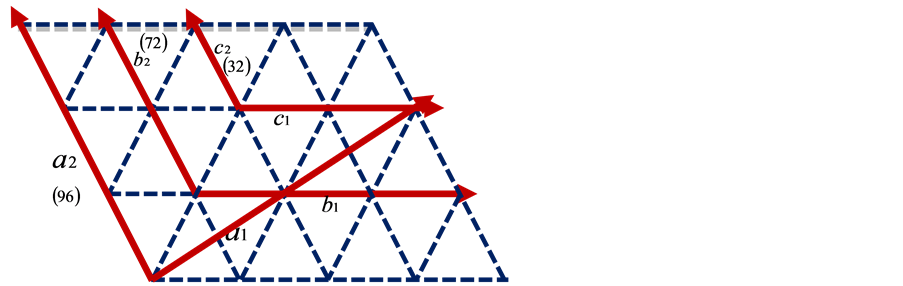
Figure 1. Top view (along [0001] direction) of the ZnO wurtzite structure: The translation vectors for the primitive unit cell and for the 32-,72-,and 96-atom supercells are shown
图1. 96原子、72原子和32原子ZnO晶体结构基矢图(俯视图,沿[0001]方向)
2.4. 缺陷跃迁能级
缺陷跃迁能级 为相同缺陷
为相同缺陷 时带电状态q和
时带电状态q和 的缺陷形成能
的缺陷形成能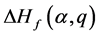 和
和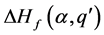 相等时的费米能 [13] 。
相等时的费米能 [13] 。
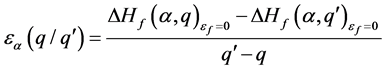 (8)
(8)
费米能级的实验值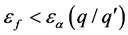 时,带电状态为q的缺陷晶体相对稳定;反之
时,带电状态为q的缺陷晶体相对稳定;反之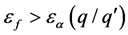 时,带电状态为
时,带电状态为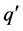 的缺陷晶体更加稳定。
的缺陷晶体更加稳定。
缺陷跃迁能级不仅能够表示晶体在某一价态时的稳定性,还能判断出晶体在室温情况下的电离性质。定义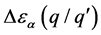 为跃迁能级与导带底或价带顶的差值为
为跃迁能级与导带底或价带顶的差值为
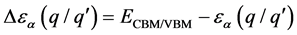 (9)
(9)
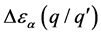 越小缺陷晶体在室温情况下越容易发生电离,称此时跃迁能级为浅施主或浅受主能级,反之为深施主或深受主能级。
越小缺陷晶体在室温情况下越容易发生电离,称此时跃迁能级为浅施主或浅受主能级,反之为深施主或深受主能级。
3. 计算结果和分析
3.1. 能带结构
使用LDA + U方法优化结构后的纤锌矿ZnO晶格参数为a = 3.149 Å,c/a = 1.604,μ = 0.382,与实验值a = 3.249 Å,c/a = 1.602,μ = 0.381 [14] 一致。结构优化后的ZnO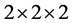 晶体及In原子替代Zn原子掺杂后结构如图2(a)和图2(b)所示。
晶体及In原子替代Zn原子掺杂后结构如图2(a)和图2(b)所示。
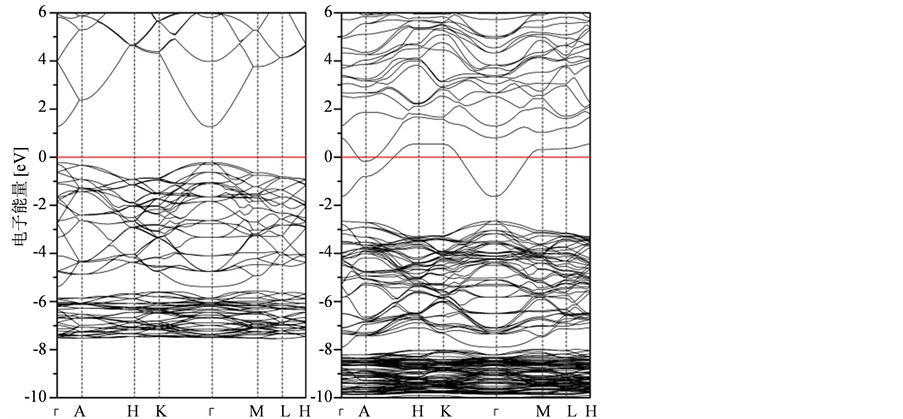
图3(a)为纤锌矿ZnO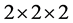 结构在布里渊区内高对称点能带结构图,纤锌矿ZnO在价带顶和导带底之间的Γ点存在一个直接带隙,LDA + U方法计算得到的直接带隙宽度为1.50 eV,仍然小于3.4 eV的实验值,但也有效改善了LDA方法计算得到的结果(0.81 eV)。
结构在布里渊区内高对称点能带结构图,纤锌矿ZnO在价带顶和导带底之间的Γ点存在一个直接带隙,LDA + U方法计算得到的直接带隙宽度为1.50 eV,仍然小于3.4 eV的实验值,但也有效改善了LDA方法计算得到的结果(0.81 eV)。
In掺杂ZnO体系能带结构图如图3(b)所示,In原子替换一个Zn原子后破坏了晶体结构本身的对称性,使得晶体的能带结构禁带宽度变小,费米能级进入导带,杂质能级靠近导带的底部,光电带隙显著增加,提高了ZnO半导体材料的光电性质。
3.2. 缺陷形成能与缺陷跃迁能级
替代掺杂情况中,在不改变96原子ZnO晶体结构参数的条件下,分别让Al、Ga和In原子在晶胞相对中间的位置替代一个Zn原子。晶格弛豫后,Al-O键、Ga-O键、In-O键键长相对于本征ZnO晶体Zn-O键键长变化量如表1所示。
由表1可以清楚的看到Al-O键和In-O键相对于Zn-O键的键长变化量大,由于Ga原子和Zn原子的半径大小接近性质相似,Ga-O键的相对变化量较小。LDA+U方法计算的ZnO的形成焓为−3.44 eV与实验值−3.58 eV [15] 接近。
LDA + U方法在计算掺杂半导体材料的能带结构时仍然会低估带隙宽度,计算的总能和形成能存在误差。但由于形成能的变化趋势是一定的,仍可根据各种掺杂结构的缺陷形成能函数图像判断其稳定性。Al、Ga、In替代Zn原子,在富锌的条件下有相对较低的形成能,掺杂结构比在富氧的条件下稳定,见图4。
富锌条件下,AlZn、GaZn、InZn掺杂均产生一个浅施主能级。AlZn和GaZn的跃迁能级相近较稳定(见表2),InZn结构因为In2O3形成后在ZnO中浓度的限制会有较高的形成能,但在三者中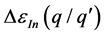 最小容易发生电离。从富锌到富氧,掺杂ZnO的缺陷形成能随着生成条件的改变逐渐增长了
最小容易发生电离。从富锌到富氧,掺杂ZnO的缺陷形成能随着生成条件的改变逐渐增长了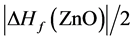 。
。

 (a)(b)
(a)(b)
Figure 2. The structure of (a) undoped ZnO 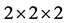 supercell and (b) In-doped ZnO
supercell and (b) In-doped ZnO  supercell
supercell
图2. (a) 本征ZnO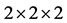 晶体结构图;(b) InZn掺杂ZnO
晶体结构图;(b) InZn掺杂ZnO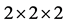 晶体结构图
晶体结构图
 (a) (b)
(a) (b)
Figure 3. Energy band structures of (a) undoped ZnO  supercell and (b) In-doped ZnO in a
supercell and (b) In-doped ZnO in a  supercell
supercell
图3. (a) 本征ZnO 晶体能带结构图;(b) InZn掺杂ZnO
晶体能带结构图;(b) InZn掺杂ZnO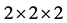 晶体能带结构图
晶体能带结构图

Table 1. The variation of Al-O, Ga-O and In-O bond lengths compared with Zn-O bond length in pure ZnO for both neutrally and positively charged states
表1. 掺杂后Al-O键、Ga-O键、In-O键键长相对于本征ZnO晶体Zn-O键键长变化量
Table 2. The transition energy levels of AlZn, GaZn and InZn after doped and the levels below the Fermi level
表2. ZnO晶体掺杂后AlZn、GaZn、InZn跃迁能级和相对于价带顶与费米能级差值
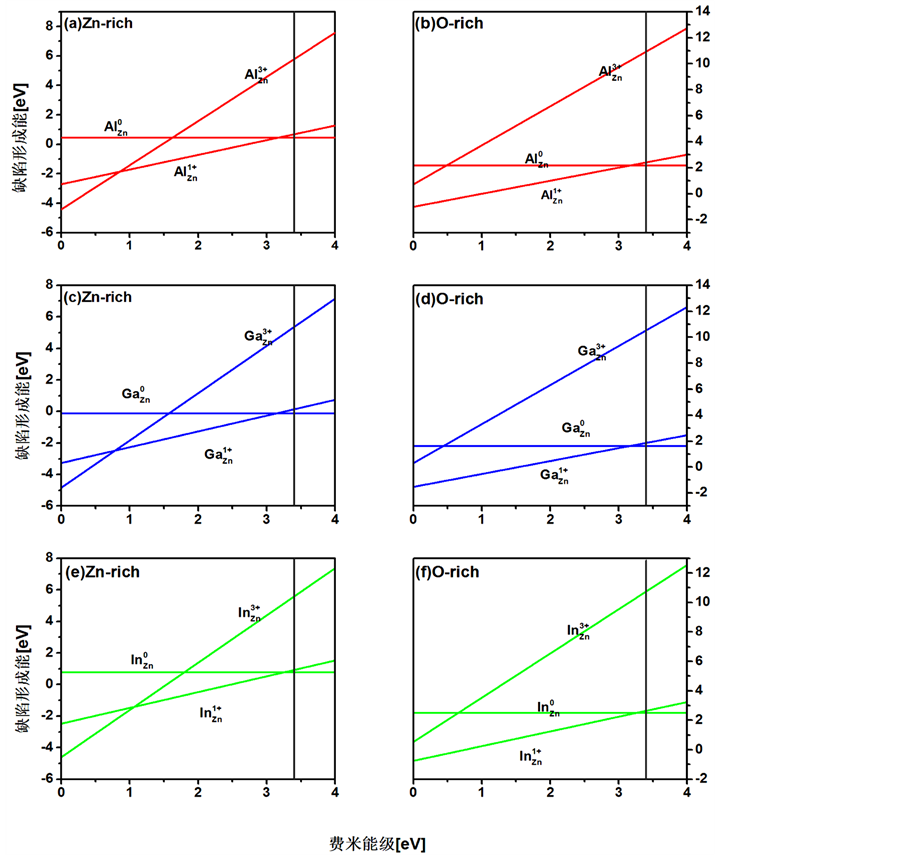
Figure 4. Formation energies as functions of Fermi level for Al, Ga, In atoms in ZnO under (a) (c) (e) zinc-rich and (b) (d) (f) oxygen-rich conditions
图4. 富Zn和富O条件下Al、Ga、In掺杂ZnO的形成能与费米能级函数图像
我们还研究了间隙掺杂ZnO超胞结构(Ali、Gai、Ini)情况,对于间隙掺杂有两种可能位置,一个是四面体间隙掺杂,另一个是八面体间隙掺杂。根据总能计算我们发现,间隙原子位于纤锌矿ZnO沿着c轴方向的六角通道中心(八面体中心)时具有最小能量。同样在富锌条件下,间隙掺杂ZnO的形成能较低,空穴可能被间隙原子补偿。而在富氧条件下,掺杂ZnO由于较高的形成能非常不易形成。Ali、Gai、Ini三种原子间隙掺杂中Gai缺陷形成能最低,Ga原子与Zn原子半径接近更容易形成稳定的掺杂晶胞。间隙掺杂原子作为施主变成+3价电状态。
4. 结论
利用LDA + U方法计算ⅢA族元素掺杂ZnO晶体光电性质,明显改善了LDA方法低估带隙宽度的问题。Al、Ga、In掺杂ZnO会在晶体中形成一个浅施主能级,In替代Zn掺杂最容易发生电离;GaZn和Gai的形成能相对较低,晶体结构更加稳定。由能带图知掺杂后的导带下移,费米能级穿过导带,理论上可以提高ZnO半导体材料的光电性质。
基金项目
黑龙江省教育厅科学技术研究项目(12511163)。
文章引用
祁雨杭,牛 丽,关 启,许华梅,卢会清,由春秋. Al、Ga、In掺杂ZnO形成能的第一性原理研究
First-Principles Studies of the Al, Ga, In-Doped ZnO Defect Formation Energy[J]. 应用物理, 2016, 06(02): 15-21. http://dx.doi.org/10.12677/APP.2016.62003
参考文献 (References)
- 1. Hong, R.J., Jiang, X., Sittinger, V., et al. (2002) Uniformity in Large Area ZnO:Al Films Prepared by Reactive Midfrequency Magnetron Sputtering. Journal of Vacuum Science & Technology A, 20, 900-905. http://dx.doi.org/10.1116/1.1472420
- 2. Kim, H.-J., Lee, H.-N., Park, J.-C., et al. (2002) The Mechanism of Im-provement of Contact Resistivity in TFT-LCDs between IZO Layers and Al-Based Metal Lines by Diffusion of Mo Atoms. Current Applied Physics, 2, 451. http://dx.doi.org/10.1016/S1567-1739(02)00097-4
- 3. Jou, J.H., Han, M.Y. and Cheng, D.J. (1992) Substrate Dependent Internal Stress in Sputtered Zinc Oxide Thin Films. Applied Physics, 71, 4333-4336. http://dx.doi.org/10.1063/1.350815
- 4. Zhou, X.H., Hu, Q.-H. and Fu, Y. (2008) First-Principles LDA+U Studies of the In-Doped ZnO Transparent Conductive Oxide. Journal of Applied Physics, 104, 063703. http://dx.doi.org/10.1063/1.2978324
- 5. Jiang, X., Wong, F.L., Fung, M.K., et al. (2003) Aluminum-Doped Zinc Oxide Films as Transparent Conductive Electrode for Organic Light-Emitting Devices. Applied Physics Letters, 83, 1875-1877. http://dx.doi.org/10.1063/1.1605805
- 6. Lu, J.G., Fujita, S., Kawaharamura, T., et al. (2007) Carrier Concentration Dependence of Band Gap Shift in n-Type ZnO:Al Films. Journal of Applied Physics, 101, 083705. http://dx.doi.org/10.1063/1.2721374
- 7. Matsubara, M., Amini, M.N., Saniz, R., Lamoen, D. and Partoens, B. (2012) Attracting Shallow Donors: Hydrogen Passivation in (Al, Ga, In)-Doped ZnO. Physical Review B, 86, 165207.
- 8. Kim, K.J. and Park, Y.R. (2001) Large and Abrupt Optical Band Gap Variation in In-Doped ZnO. Applied Physics Letters, 78, 475-477. http://dx.doi.org/10.1063/1.1342042
- 9. Ye, J.D., Gu, S.L., Zhu, S.M., Liu, S.M., Zheng, Y.D., Zhang, R. and Shi, Y. (2005) Fermi-Level Band Filling and Band-Gap Renormalization in Ga-Doped ZnO. Applied Physics Letters, 86, 192111. http://dx.doi.org/10.1063/1.1928322
- 10. Paier, J., Hirschl, R., Marsman, M. and Kresse, G. (2005) The Per-dew-Burke-Ernzerhof Exchange-Correlation Functional Applied to the G2-1 Test Set Using a Plane-Wave Basis Set. Journal of Chemical Physics, 122, 460-470. http://dx.doi.org/10.1063/1.1926272
- 11. Sanizn, R., Xu, Y., Matsubara, M., Amini, M.N., Dixit, H., Lamoen, D. and Partoens, B. (2013) A Simplified Approach to the Band Gap Correction of Defect Formation Energies: Al, Ga, and In-Doped ZnO. Journal of Physics and Chemistry of Solids, 74, 45-50. http://dx.doi.org/10.1016/j.jpcs.2012.07.017
- 12. Van de Walle, C.G. and Neugebauer, J. (2004) First-Principles Calculations for Defects and Impurities: Applications to Ⅲ-Nitrides. Journal of Applied Physics, 95, 3851-3879. http://dx.doi.org/10.1063/1.1682673
- 13. Janotti, A. and Van de Walle, C.G. (2007) Native Point Defects in ZnO. Physical Review B, 76, 165202. http://dx.doi.org/10.1103/PhysRevB.76.165202
- 14. Thomas, M. (1997) Semiconductors-Basic Data. 2nd ed. Edited by O. Madelung, Springer, Berlin, 1996, viii, 317 pp., hardcover, DM 88.00, ISBN 3-540-60883-4. Chemical Vapor Deposition, 3, 288-289.
- 15. Dean, J.A. (1992) Book Review: Lange’s Handbook of Chemistry. 14th Edition, McGraw-Hill, New York.
*通讯作者。