Pharmacy Information
Vol.04 No.01(2015), Article ID:14770,13
pages
10.12677/PI.2015.41001
The Quality Control of the Malaridine Phosphate Capsule
Hongjie Wang
Great Biology Pharmaceutical (Tianjin) Co. Ltd., Tianjin
Email: 2006wanghongjie@163.com
Received: Jan. 13th, 2015; accepted: Jan. 23rd, 2015; published: Jan. 30th, 2015
Copyright © 2015 by author and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Malaria is an infectious disease which is spread by mosquitoes biting. Its features are shiver, high fever, sweating defervescence, anemia and splenomegalia. Malaria remains one of the ten largest infectious diseases globally. According to statistics, 300 - 500 million people are infected by malaria and 1 million people are died of malaria annually. The new anti-malarial medicine synthesized at the beginning of 1970s exists in the form of tetraphosphate clinically. This paper is to determine the prescription and production process of new drug—Malaridine Phosphate Capsule, establish quality standards, perform pilot scale production and detection, carry out acceleration and long-term stability investigation and research pharmacology, toxicology, pharmaceutical effect, pharmacokinetics, etc. It is to determine the optimum prescription and process by orthogonal test, hydromechanics, angle of repose research and pharmaceutical technology. It is to build quality-control method by HPLC and UV; to perform pilot large-scale production and detection in national GMP workshop, detect the appearance, related substances, dissolution rate and content of commercially available packages by sampling in 40˚C, 40% relative humidity and 25% ± 5%, 60% ± 5% relative humidity respectively according to new drug stability guiding principle. Conclusion: 1) Reasonable prescription. 2) Simple and feasible process. 3) Quality standards and detection method have been established. 4) 2-year of stable-quality expiry date. 5) Obvious economical and social benefits instead of inadequate conventional formulations and bioavailability.
Keywords:Malaridine Phosphate Capsule, Prescription, Craft, Stability
磷酸咯萘啶胶囊的质量研究
王洪杰
格特生物制药(天津)有限公司,天津
Email: 2006wanghongjie@163.com
收稿日期:2015年1月13日;录用日期:2015年1月23日;发布日期:2015年1月30日

摘 要
疟疾(malaria)是由疟原虫经按蚊叮咬传播的污染病。临床上以周期性定时性发作的寒战、高热、出汗退热及贫血和脾大为特点。疟疾仍然是国际上十大传染病之一,据统计每年发病人数为3~5亿,死于疟疾者约百万人。咯萘啶是70年代初合成抗疟新药,临床上主要以四磷酸盐形式存在。本文确定磷酸咯萘啶胶囊新药处方、生产工艺、建立质量标准、中试生产并检测、进行加速和长期稳定性考察、研究药理毒理药效药代学等。采用正交试验法、流体力学、休止角研究和制药技术确定最佳处方及工艺,建立了HPLC和UV进行控制质量方法;在国家GMP车间内进行中试规模化生产并检测,采用市售包装于
关键词 :磷酸咯萘啶胶囊,处方,工艺,稳定性

1. 引言
疟疾分布广泛,北纬60˚至南纬30˚之间,海拔
2. 处方及工艺筛选及分析
2.1. 处方筛选
处方设计中分别考察了填充剂种类,粘合剂种类,崩解剂种类、润湿剂种类及相关用法对生产工艺及产品质量的影响[5] [6] ,见表1。
根据常规制剂胶囊组方中需要主药和填充剂,制备过程需要制粒、干燥、整粒、装囊等工艺规程,检测其成形、颗粒流动性、颗粒崩解性、颗粒溶解性、溶出度等中控指标,进行处方筛选和工艺考核。其中处方八组方合理,工艺最佳,因此选处方八为最终处方。

Table 1. Prescription and process selection
表1. 处方及工艺筛选
2.2. 工艺筛选
2.2.1. 处方分析
处方一:颗粒不成形,可能与淀粉少并且微晶纤维素松散有关;
处方二:成形,是在处方一基础上加减和更改辅料;
处方三:成形崩解度差,由于粘合剂淀粉浆加入量太大有关;
处方四:不成形,与淀粉量大,粘合剂淀粉浆少,不能完全粘合在一起有关;
处方五:是在处方三基础上加减和更改辅料;
处方六:成形不崩解,由于粘合剂淀粉浆加入量太大有关;
处方七:根据常规大众制备法,淀粉外加和内加法制备颗粒,不加硬脂酸镁做润滑剂,结果颗粒流动性不乐观。
处方八:组方合理,工艺最佳。
处方九:雷同处方三;
处方十:粘度大,制软材时采用粘合剂淀粉浆一次加入法,所以粘度大,而处方八处方基本相似,软材制备过程采用等量递增逐步添加粘合剂淀粉浆的方法粘度适宜,成形性好;
处方十一:粘度小不成形,粘合剂剂淀粉浆加入量太小有关,采用水做润湿剂后,粘度加大,仍不成形;
处方十二:成形崩解度好工艺繁琐成本高,但组方中辅料繁多,制备工艺复杂,成本高,不符合工业化大生产的需要。
2.2.2. 检测方法
1) 崩解时限检查法:《中国药典》2010年版二部附录83页XA崩解时限检查法,取供试品6粒进行崩解时限检查,应在30分钟内全部崩解[7] 。
2) 休止角考察方法:《药剂学》第三版(人民卫生出版社出版),休止角测定法1 [8] 。
3) 稳定性试验:《中国药典》2010年版二部附录199页XIXC药物制剂稳定性试验指导原则,加速试验按市售包装,放入相对湿度75% ± 5%的恒温箱(温度40℃),分别于第1、2、3和6个月末取样一次,按稳定性试验重点考察项目进行检测,与0个月考察结果比较。长期稳定性试验按市售包装,于恒温恒湿环境(温度
2.3. 制备工艺
a) 将处方量磷酸咯萘啶、淀粉、蔗糖、硬脂酸镁分别过80目筛备用,淀粉配成10%淀粉浆备用。
b) 称取处方量磷酸咯萘啶、淀粉、蔗糖各组分,等量递加,混匀。
c) 向粉末混合物中加入10%淀粉浆搅拌20 min制成软材,16目筛制粒。
d) 58℃ ±
e) 12目筛整粒,颗粒加入处方量的硬脂酸镁,混匀(测定颗粒含量控制在98.0%~102.0%)。
f) 装4#囊。
g) 铝塑泡罩包装。
h) 合格品包装,入库。
3. 质量研究及分析
3.1. 试验材料及设备
3.1.1. 原辅料来源
原辅料名称 批号 生产厂家
磷酸咯萘啶012002 浙江民生制药厂
淀粉 20130301 曲阜市药用辅料有限公司
微晶纤维素 20130316 曲阜市药用辅料有限公司
蔗糖 20130308 曲阜市药用辅料有限公司
硬脂酸镁 20130106 曲阜市药用辅料有限公司
胶囊 20130203 浙江绿键胶囊有限公司
铝箔 20120304 上海盛装包装材料有限公司
聚氯乙烯固体药用硬片 20120108 济南兰光机电技术有限公司
3.1.2. 对照品及试剂
原辅料名称 批号 生产厂家
磷酸咯萘啶对照品 H01203 中国生物制品检定所
磷酸二氢钾 20120406 天津市大茂化工有限公司
乙腈 20121001 天津市四友精细化学品有限公司
甲醇 20120916 天津市四友精细化学品有限公司
磷酸 20120406 天津市大茂化工有限公司
盐酸 20120311 天津市大茂化工有限公司
3.1.3. 检验仪器
设备名称 型号 生产厂家
酸度计 FE20型 梅特勒–托利仪器有限公司
电热恒温干燥箱 202-1AB型 天津泰斯特仪器有限公司
电子天平 PCB600-2型德国KERN公司
电子天平 AL204型 梅特勒–托利仪器有限公司
电子分析天平 AB265-S型 梅特勒–托利仪器有限公司
电热恒温水浴锅 DZKW-4型 北京中兴伟业仪器有限公司
电热真空干燥箱 DZF-2型 北京永光明医疗仪器厂
手提式压力蒸汽消毒器 GMSX-280型 北京永光明医疗仪器厂
电热恒温培养箱 DH4000B型 天津泰斯特仪器有限公司
智能程序马弗炉 XMT-908型 余姚市长江温度仪表厂
澄明度检测仪 YB-Ⅱ型 天津天光光学仪器有限公司
生化培养箱 SHP-250型 北京中兴伟业仪器有限公司
熔点仪 YRT-3型 天津市天大天发科技有限公司
三用紫外分析仪 ZF-2型 上海安亭电子仪器厂
电导率仪 DDS-307型 上海康仪仪器有限公司
洁净工作台 CJ-820型 苏州苏信净化设备厂
数控超声清洗器 KQ-50DE型 昆山舒美超声仪器有限公司
磁力搅拌器 90-4型 上海振捷实验设备有限公司
紫外可见分光光度计 T6型 北京普析通用有限责任公司
水分滴定仪 ZSD-1型 上海安亭电子仪器厂
高效液相色谱仪 LC系列 日本岛津
3.2. 有关物质试验研究
3.2.1. 测定方法的拟定
仪器:岛津高效液相色谱仪系统。LC-10ATVP泵/SPD-10AVP紫外检测器/分析之星工作站;六通阀进样器(20 ml定量进样环)。
测定法:取本品研细,精密称取细粉(相当于磷酸咯萘啶100 mg)置50 ml容量瓶中,用水溶解并稀释至刻度,滤过,取续滤液5.0 ml至50.0 ml的容量瓶中,用流动相稀释至刻度,滤膜过滤,续滤液作为供试溶液。精密量取适量,用流动相稀释,制成20 ug/ml的溶液,作为对照溶液,量取对照溶液20 ul进样,调节监测器灵敏度,使主成分色谱峰高为满量程的20%。精密量取供试溶液20 ul,注入色谱仪,记录色谱图至主峰保留时间的2倍,供试溶液色谱图中如有杂质峰,量取各杂质峰面积的和,以对照溶液的峰面积为对照,计算杂质总量。
3.2.2. 色谱条件的选择
1) 色谱柱:选择反相C18色谱柱(250 mm ´ 4.6 mm,5 mm),即十八烷基硅烷键合硅胶为填充剂的色谱柱。
2) 测定波长:检测波长为260 nm和276 nm波长处有最大吸收,选择276 nm作为本品有关物质检查用检测波长。
3.2.3. 色谱条件的拟定
固定相:以十八烷基硅烷键合硅胶为填充剂的色谱柱。
流动相:0.08 mol/L磷酸二氢钾(pH2.8):乙腈:甲醇 = 79:6:15。
检测波长:276 nm。
流速:1.0 ml/min。
3.2.4. 系统适用性试验
取磷酸咯萘啶胶囊适量,用流动相溶解并稀释制成0.2 mg/ml的溶液,在上述色谱体系下,进样20 µl,注入液相色谱仪,记录色谱图,结果主峰与邻近杂峰分离度R = 2(12.62 − 10.49)/1.8296 = 2.33,理论塔板数以主峰计算n = 5.54(12.62/0.448)2 = 4396,磷酸咯萘啶峰与其他峰可完全分离,理论板数超过2000。结果:分离度(R)为2.33,理论板数(N)为4396。
3.2.5. 专属性试验
溶剂:取水,按上述色谱条件,进样20 μl,记录色谱图,结果没有显著影响。
辅料:取辅料混合物50.0 mg,精密称定,至100 ml量瓶中,加适量水,振摇,加水稀释至刻度,滤过,弃去初滤液,取续滤液5.0 ml于50 ml的容量瓶中,用流动相稀释至刻度,滤膜过滤,取续滤液20 μl注入色谱仪,记录色谱图,结果显示辅料对本品的有关物质测定没有显著影响。
3.2.6. 保留时间的设定
经过上述方法学的研究,流速为1.0 ml/min时,磷酸咯萘啶13 min左右出峰,样品及破坏后的样品在26 min以后未见明显杂质峰出现,因此,设定记录色谱图至磷酸咯萘啶峰保留时间的3倍。
3.2.7. 测定方法的选择
选择面积归一化法测定供试品中有关物质的含量。
3.2.8. 三批样品有关物质测定
按所建方法对三批样品进行磷酸咯萘啶有关物质检查,结果见表2。
结论:三批样品中有关物质均 < 1.0%。
3.3. 磷酸咯萘啶胶囊溶出度试验研究
3.3.1. 方法的专属性
取胶囊空白辅料的混合物约17 mg~18 mg,置100 ml量瓶中,用0.01 mol/L的盐酸溶解并稀释至刻度,摇匀,滤过,精取续滤液10.0 ml,置100 ml量瓶中,用0.01 mol/L的盐酸稀释至刻度,照分光光度法在200~400 nm扫描检测,并在276 nm处测定吸收度,结果显示混合物在276 nm处几无吸收,对磷酸咯萘啶的溶出度测定无干扰。
3.3.2. 波长的选择
精密称取磷酸咯萘啶(约10 mg),用0.01 mol/L的盐酸溶解溶解并稀释制成10 µg/ml的溶液,在200~ 400 nm波长处进行波长扫描,在260 nm和276 nm波长处有最大吸收,选择276 nm为检测波长。
3.3.3. 线性范围
精密称取
Table 2. Results of three batches of sample determination of related substances
表2. 三批样品有关物质测定结果
别为0.5 mg/L、1 mg/L、2 mg/L、4 mg/L、6 mg/L、8 mg/L、10 mg/L和15 mg/L的溶液。照紫外–可见分光光度法在276 nm的波长处分别测定吸收度,以浓度C(μg/ml)为横坐标,吸收度A为纵坐标,按最小二乘法计算回归方程与相关系数。结果:见表3,标准曲线见图1。
磷酸咯萘啶在0.4~14.9 mg/L的范围内线性关系良好,线性方程为y = 0.059x + 0.0069,相关系数r = 0.9996。
3.3.4. 回收率试验
测定方法:取
分光光度法60%~120%的平均回收率为99.70%,RSD = 0.01%。溶出测定方法的回收率在98.0%~102.0%之间,符合要求。
3.3.5. 精密度试验
取回收率试验100%测定项下溶液,将样品溶液重复测定6次,结果如下:试验结果表明溶液稳定性
Table 3. Result of linear range test
表3. 线性范围试验结果
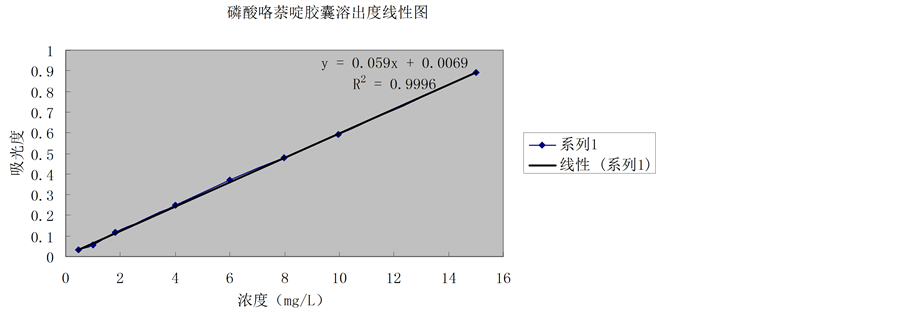
Figure 1. The linear degrees of dissolution of pyronaridine phosphate capsules
图1. 磷酸咯萘啶胶囊溶出度线性
较好,RSD = 0.01% (N = 6),可满足定量分析的要求。结果见表5。
3.3.6. 溶出方法的建立
参考溶出度测定法(《中国药典》2010年版二部附录85页XC溶出度测定法第一法篮法和第二法浆法),以0.01 mol/L盐酸溶液900 ml为溶剂,研究磷酸咯萘啶胶囊在不同溶出条件下的溶出情况,以确定最佳的溶出方法。
1) 溶出体积的选择
参考溶出度测定法以0.01 mol/L盐酸溶液为溶剂,溶出体积为900 ml,考虑到UV测定浓度的问题,故采用900 ml为溶出体积。
2) 溶出方法的选择
照溶出度测定法以0.01 mol/L盐酸溶液900 ml为溶剂,第一法转速为50转/分钟;第二法转速也为50转/分钟。依法操作,经5、10、15、30、45、60分钟取样5.0 ml,微孔滤膜滤过,精密吸取续滤液0.5 ml,置于10 ml容量瓶中,用0.01 mol/L盐酸溶液稀释至刻度,作为供试品溶液;另精密称取
Table 4. Results of recovery test
表4. 回收率试验结果
Table 5. Test results of precision
表5. 精密度试验结果
溶液。分别取供试品溶液和对照品溶液,紫外–可见分光光度法分别于276 nm波长处,以盐酸(0.01 mol/L)的溶液作为空白溶液,测定供试品溶液和对照品溶液的吸收度,计算每粒胶囊中主要成分的溶出量。照上述拟定的磷酸咯萘啶胶囊溶出度测定方法,测定磷酸咯萘啶在不同时间的溶出量,绘制溶出曲线。结果见表6,图2。
从测定结果看,浆法的溶出度比篮法溶出度高,因此选定第二法浆法为溶出度测定方法。
3) 取样时间的确定
本品溶出曲线显示,在上述条件下(以水为溶剂,浆法,转速为每分钟50转),30 min时的溶出量均大于80.0%,其后溶出曲线已趋于平缓,且溶出均一性均较好,故选择30 min作为取样时间。
Table 6. Pyronaridine phosphate capsules paddle method, basket dissolution curves of 1
表6. 磷酸咯萘啶胶囊桨法、篮法溶出度曲线1

Figure 2. Pyronaridine phosphate capsule second method (slurry method) and the first method (basket) dissolution curve
图2. 磷酸咯萘啶胶囊第二法(浆法)及第一法(篮法)溶出度曲线
4) 自制磷酸咯萘啶胶囊与市售磷酸咯萘啶片在人工肠液中的溶出比较
测定方法:照溶出度测定法以人工肠液900.0 ml为溶出介质,转速为每分钟50转;依法操作,胶囊于5、10、15、30、45、60、75分钟取样5 ml,片剂于10、15、30、45、60、75分钟取样5.0 ml,微孔滤膜滤过,精密吸取续滤液0.5 ml,置于10 ml容量瓶中,用盐酸(0.01 mol/L)稀释至刻度,作为供试品溶液,依法测定。结果见表7,图3。
试验结果表明:采用定量分析法比较,自制的磷酸咯萘啶胶囊比市售磷酸咯萘啶片溶出速度快。
3.3.7. 均一性考察
按照确定的溶出条件和方法,测定三批样品在不同时间的溶出量,并绘制溶出曲线。结果见表8;溶出曲线见图4。
Table 7. Homemade pyronaridine phosphate capsules with commercially available malaridine phosphate tablets in artificial intestinal juice dissolution
表7. 自制磷酸咯萘啶胶囊与市售磷酸咯萘啶片在人工肠液中的溶出度
Table 8. The dissolution of three batches of samples
表8. 三批样品溶出度
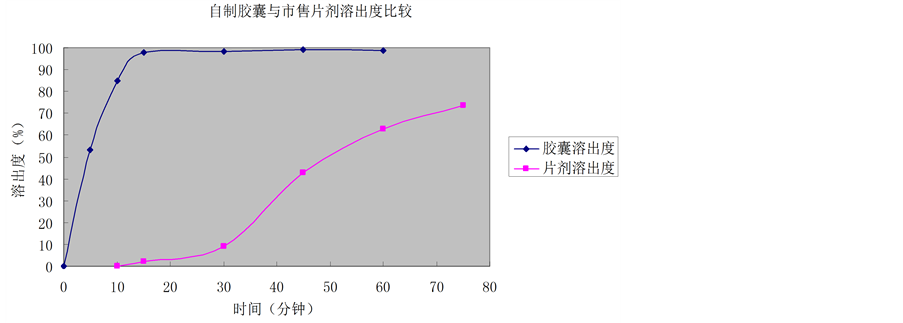
Figure 3. Homemade pyronaridine phosphate capsules with commercially available malaridine phosphate tablets in artificial intestinal juice dissolution curve
图3. 自制磷酸咯萘啶胶囊与市售磷酸咯萘啶片在人工肠液中的溶出曲线
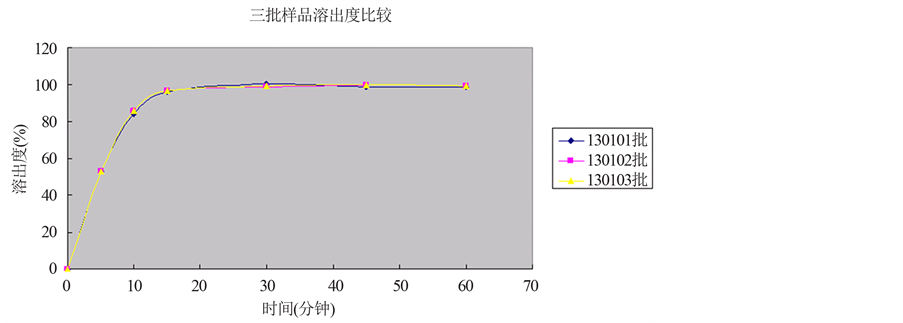
Figure 4. Dissolution curves of the three batch of malaridine phosphate capsules slurry method
图4. 三批磷酸咯萘啶胶囊浆法溶出曲线
结论:三批样品溶出均一性较好,5分钟时三批样品溶出度为53%左右,10分钟时三批样品溶出度为85%左右,15分钟时三批样品溶出度为96%左右,30分钟时三批样品溶出度为99%左右,45分钟时三批样品溶出度为99%左右,60分钟时三批样品溶出度为99%左右,各个时间段三批样品溶出度较一致,各辅料迅速崩解使有效成分迅速释放并达到较高浓度,15分钟内该药物的溶出度呈明显上升趋势,15分钟之后溶出度曲线呈平滑状态,达到最大溶出度,三批样品的溶出度均一性较好。
3.4. 含量方法学研究
3.4.1. 测定方法的拟定
对照品溶液的制备:精密称取
供试品溶液的制备: 取本品10粒,除去囊壳,研细,精密称取适量(约相当于磷酸咯萘啶10 mg),置100 ml棕色容量瓶中,加入磷酸盐缓冲液(pH 7.0)适量使磷酸咯萘啶溶解并稀释至刻度,3000转/分离心10分钟,取上清作为供试品溶液。
测定法 精密量取对照品溶液与供试液各5.0 ml,分别置50 ml棕色容量瓶中,用磷酸盐缓冲液(pH 7.0)稀释至刻度,摇匀,紫外–可见分光光度法在276 nm的波长处分别测定吸收度,计算,并将计算结果乘于0.569,即得供试液中含有C29H32ClN5O2的量。
3.4.2. 测定波长的选择
取磷酸咯萘啶适量(约10 mg),磷酸盐缓冲液(pH 7.0)溶解并稀释制成10 µg/ml的溶液,在200~400 nm下扫描,在260 nm和276 nm处有最大吸收,选择276 nm为检测波长。
3.4.3. 线性范围
方法:对照品溶液的制备:精密称取
测定方法:分别取对照品储备,用磷酸盐缓冲液(pH 7.0)制成0.5、1、2、4、6、8、10、15 mg/L的溶液。照紫外分光光度法在276 nm的波长处分别测定吸收度(以磷酸盐缓冲液(pH 7.0)为空白),按拟定的含量方法测定,以浓度C(μg/ml)为横坐标,峰面积A为纵坐标,按最小二乘法计算回归方程与相关系数。标准曲线见图5。
线性研究表明:磷酸咯萘啶在0.5~15 mg/L的范围内有良好的线性关系,线性方程为y = 0.0322x + 0.012,r = 0.9996。
3.4.4. 进样精密度
取回收测定项下同一样品溶液,重复测定6次,连续进样6次,结果见表9。
结论:该法进样精密度较好,符合测试要求。
3.4.5. 回收率试验
精密称取
从上面结果看回收率较低,回收的RSD较大,因此我们将测定方法进行改进,用离心(3500转/分离心15分钟)的方法代替滤纸过滤的方法处理样品。回收结果如下:
结论:本法80%~120%的平均回收率,磷酸咯萘啶为99.88%,RSD = 0.09%;磷酸咯萘啶的回收率均在98.0%~102.0%之间,符合要求。
3.4.6. 重复性
按拟定的含量测定方法,取样品(批号:130201)6份,测定每粒中磷酸咯萘啶的含量。结果见表12。
结论:本法重复性良好。
3.4.7. 三批样品检测
照拟定的含量测定方法测定中试三批样品磷酸咯萘啶胶囊的含量,结果见表13。

Figure 5. Malaridine phosphate content in a linear standard curve
图5. 磷酸咯萘啶含量线性标准曲线
Table 9. Sample results of precision test table
表9. 进样精密度试验结果表
Table 10. Pyronaridine phosphate results of recovery test
表10. 磷酸咯萘啶回收率试验结果
Table 11. Pyronaridine phosphate results of recovery test
表11. 磷酸咯萘啶回收率试验结果
Table 12. Pyronaridine phosphate repeatability test results (n = 6)
表12. 磷酸咯萘啶重复性试验结果(n = 6)
Table 13. The three batch of samples of pyronaridine phosphate capsule content determination results
表13. 三批样品磷酸咯萘啶胶囊含量测定结果
4. 小结
磷酸咯萘啶胶囊(C29H32ClN5O2)经处方筛选、工艺考核、有关物质、溶出度和含量质量方法学研究,该制备方法处方合理工艺最佳,检测方法快速、简便、准确、灵敏度高,便于生产企业规模化生产和检测,该方法具有研究性的意义和研究前瞻。
文章引用
王洪杰, (2015) 磷酸咯萘啶胶囊的质量研究
The Quality Control of the Malaridine Phosphate Capsule. 药物资讯,01,1-14. doi: 10.12677/PI.2015.41001
参考文献 (References)
- 1. 丁雪 (2008) 21中国疟疾流行现状综述. 中华流行病学杂志, 2, 112-113.
- 2. Cheng, C. and Zheng, X.Y. (1992) Development of the new antimalarial pyronaridine: A review. Biomedical and Environmental Sciences, 5, 149-160.
- 3. Pascal, R., Jean, B. and Leonardo, K. (1996) BASCO in vitro activity of antimalarials clinical isolates of plasmodium falciparum in Yaounde. The American Journal of Tropical Medicine and Hygiene, 55, 254-258.
- 4. Ringwald, P., Bickii, J., Same-Ekobo, A. and Basco, L.K. (1997) Pyronaridine for Treatment of Plas-modium ovale and Plasmodium malariae Infections. Antimicrobial Agents and Chemotherapy, 41, 2317-2319.
- 5. 上海医药工业研究院药物制剂部、药物制剂国家工程研究中心编著 (2002) 药物辅料应用技术. 第二版, 中国医药科技出版社, 北京.
- 6. 罗明生主编 (1995) 药剂辅料大全. 四川科学技术出版社, 成都.
- 7. (2010) 中国药典2010年版二部.
- 8. 奚念朱主编 (1980) 药剂学. 第三版, 人民卫生出版社出版, 北京, 休止角测定法1.