 Material Sciences 材料科学, 2013, 3, 199-205 http://dx.doi.org/10.12677/ms.2013.35036 Published Online September 2013 (http://www.hanspub.org/journal/ms.html) Theoretical Study on the Interaction between the M(II)/Al-LDH(M=Zn, Mg) Layer and the Interlayer Anion 1-Anilinonaphthalene-8-Sulfonate (1,8-ANS)* Chongyang Zhao, Hong Yan# School of Science, Beijing University of Chemical Technology, Beijing Email: nagasewa@sina.com.cn, #yanhong@mail.buct.edu.cn Received: Jun. 26th, 2013; revised: Jul. 19th, 2013; accepted: Jul. 30th, 2013 Copyright © 2013 Chongyang Zhao, Hong Yan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract: The MgAl or ZnAl layered double hydroxide (LDHs) layer cluster and 1,8-ANS were optimized by density functional theory (DFT) at the level of B3LYP-6-31G(d,p)//Lanl2DZ. The molecular electronic potential (MEP) of the LDHs clusters and 1,8-ANS are also investigated. The interaction energies between 1,8-ANS and MgAl or ZnAl LDHs layer were calculated. The results indicate that the interaction between 1,8-ANS and MgAl LDHs layer is slightly stronger than that between 1,8-ANS and ZnAl LDHs layer. However, 1,8-ANS and ZnAl LDHs layer is more stable than 1,8-ANS and MgAl LDHs layer in the view of energy. Keywords: Layered Double Hydroxide (LDH); 1-Anilinonaphthalene-8-Sulfonate (1,8-ANS); Interaction Energy; Density Functional Theory; Molecular Dynamics M(II)/Al-LDH(M=Zn, Mg)层板与 1,8-ANS 客体分子相互作用的理论研究* 赵重阳,鄢 红# 北京化工大学理学院,北京 Email: nagasewa@sina.com.cn, #yanhong@mail.buct.edu.cn 收稿日期:2013 年6月26 日;修回日期:2013年7月19 日;录用日期:2013 年7月30日 摘 要:利用密度泛函理论,在B3LYP-6-31G(d,p)//Lanl2DZ 水平上分别优化了有机荧光分子 8-苯胺-1-萘磺酸 (1,8-ANS)与镁铝、锌铝水滑石层板单体及1,8-ANS 与两团簇复合体的几何结构,计算分析了两者静电势分布以 及相互作用能的大小。结果表明 1,8-ANS 与镁铝水滑石层板之间相互作用较之与锌铝水滑石层板之间相互作用 更大,从总体能量上看,锌铝-ANS水滑石体系较镁铝-ANS体系更为稳定。 关键词:类水滑石(LDHs);1,8-ANS;相互作用能;密度泛函理论研究;分子动力学 1. 引言 有机荧光染料1,8-ANS 的荧光性质(荧光波长,荧 光强度以及量子产率等)对环境非常敏感,因而作为荧 光探针广泛用于测定蛋白质折叠[1,2],测定微环境疏水 亲水性[3],测定蛋白质活性结合位点[4]等领域。但在 溶液中 1,8-ANS的荧光易发生不同程度的猝灭[5]。将 其吸附到极性固体基质上是常用的减少荧光猝灭的 有效手段[6]。 *基金项目:国家自然科学基金(21101011)。 #通讯作者。 Copyright © 2013 Hanspub 199  M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 水滑石材料是一种阴离子型层状化合物,它具有 夹层状的结构并具有良好的层间离子交换性[7],已在 催化、分子容器、纳米材料等诸多领域引起研究者的 广泛关注[8-10]。最近,实验中将水滑石作为基质,将 有机荧光染料插到水滑层间形成固体复合荧光材料, 大大提高了其发光效率。实验结果表明,1,8-ANS 引 入到锌铝水滑石中能发出比其被引入到镁铝水滑石 强的荧光[11]。这说明,1,8-ANS 与锌铝,镁铝水滑石 层板之间的相互作用存在一定的差异。本文旨在通过 量子化学理论计算从微观层次上解释、澄清不同层板 与1,8-ANS 的相互作用。 本文利用密度泛函理论,分别优化了1,8-ANS 与 锌铝、镁铝水滑石团簇以及1,8-ANS 单体与二者复合 体团簇的几何结构,分析了两者静电势分布以及相互 作用能的大小。同时利用分子动力学方法计算了 1,8- ANS 在镁铝、锌铝水滑石层间的排布方式。 2. 计算模型与计算方法 2.1. 1,8-ANS单体的计算 单个 1,8-ANS( )分子的优化采用的计 算模型如图 1。图3~5、7、9原子标号和原子种类的 配色方案均与图1、图 2相同。 16 123 CHNSO Figure 1. Structure of 1,8-ANS 图1. 1,8-ANS分子结构图 Figure 2. Model of the cluster of M/Al-LDH(M=Mg, Zn) layer 图2. 锌铝、镁铝水滑石层板团簇模型 2.2. 水滑石层板团簇的优化 根据我们以前的研究,三个原子的团簇是既能反 映层板的几何结构,又最省时的模型[12],故建立如下 锌铝,镁铝水滑石团簇的模型。 2.3. 1,8-ANS在水滑石层间的排布形式 用ONIOM[13]方法计算 1,8-ANS 在层板间的取 向。将 1,8-ANS分子作为量化层(QM 层),层板结构 应用 UFF 力场,用分子动力学方法进行模拟。因层板 带正电荷,1,8-ANS 的磺酸基部分带负电荷较为集中, 故构建模型如图3。水滑石层板结构模型以Mg/Al 水 滑石结构(空间群 R-3 m)为基础,分别构建 Mg/Al- LDHs 和Zn/Al-LDHs 6 × 6 × 1 的菱形超晶胞[14]。计算 时几何参数保持不变。 2.4. 1,8-ANS与水滑石团簇相互作用能的计算 结合水滑石团簇中各原子的电荷密度以及 ONIOM 方法优化得到 ANS 与水滑石层板的相对位置,建立 了水滑石团簇与1,8-ANS 复合体的模型,如图 4。在 对复合物体系优化过程中,由于描述的具有相互作用 的两个体系的基函数轨道有所重叠,因此进行了基组 Figure 3. The model of the simulation for orientation of ANS in the M/Al-LDH(M=Mg, Zn) interlayers 图3. ANS与镁铝、锌铝水滑石取向模拟的初始模型 Figure 4. The model of complex of 1,8-ANS and Zn(Mg)Al cluster 图4. 锌(镁)铝水滑石层板团簇与 1,8-ANS 复合体模型 Copyright © 2013 Hanspub 200 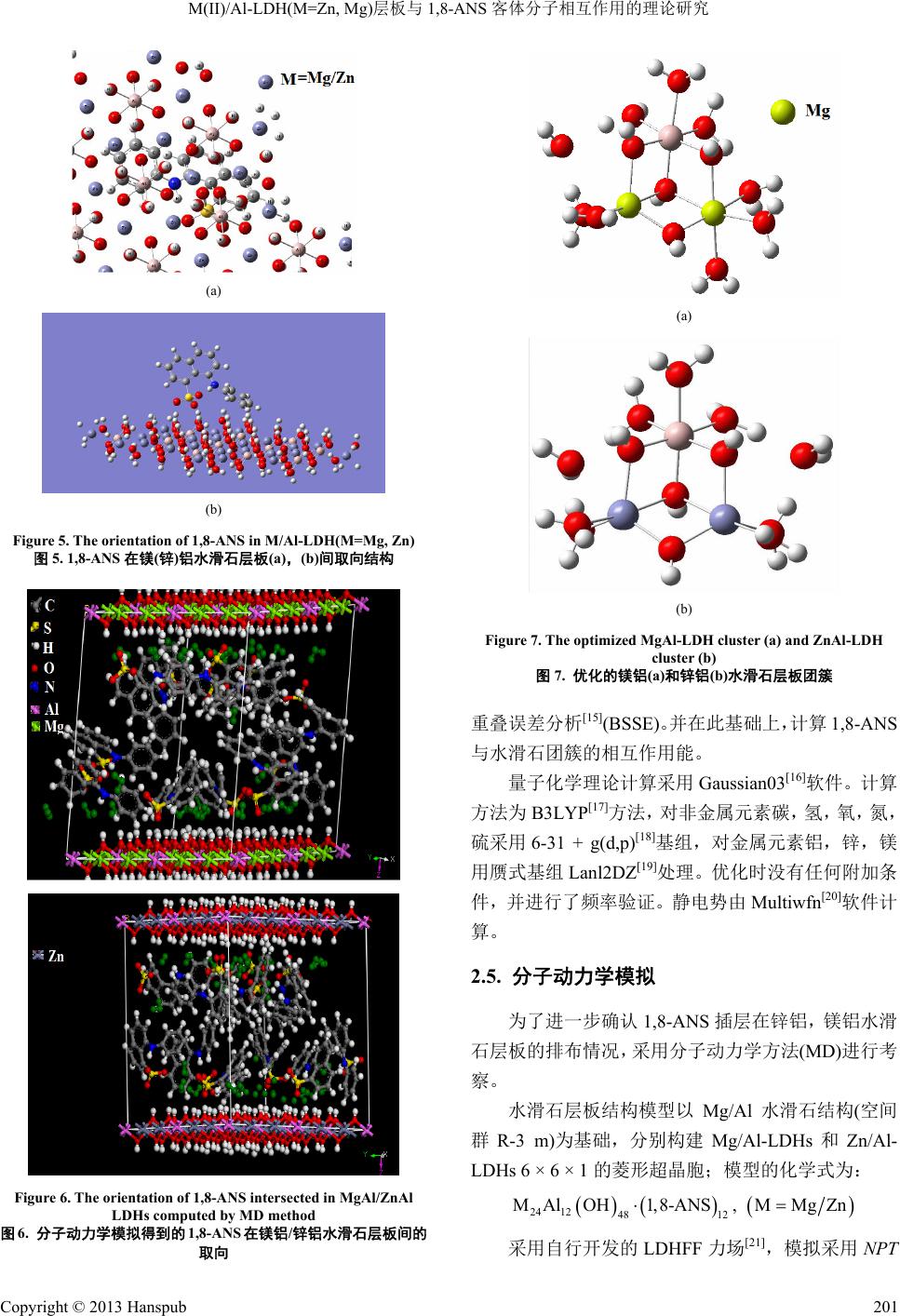 M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 (a) (b) Figure 5. The orientation of 1,8-ANS in M/Al-LDH(M=Mg, Zn) 图5. 1,8-ANS在镁(锌)铝水滑石层板(a),(b)间取向结构 Figure 6. The orientation of 1,8-ANS intersected in MgAl/ZnAl LDHs computed by MD method 图6. 分子动力学模拟得到的 1,8-ANS在镁铝/锌铝水滑石层板间的 取向 (a) (b) Figure 7. The optimized MgAl-LDH cluster (a) and ZnAl-LDH cluster (b) 图7. 优化的镁铝(a)和锌铝(b)水滑石层板团簇 重叠误差分析[15](BSSE)。并在此基础上,计算 1,8-ANS 与水滑石团簇的相互作用能。 量子化学理论计算采用Gaussian03[16]软件。计算 方法为 B3LYP[17]方法,对非金属元素碳,氢,氧,氮, 硫采用 6-31 + g(d,p)[18]基组,对金属元素铝,锌,镁 用赝式基组Lan l2DZ[19]处理。优化时没有任何附加条 件,并进行了频率验证。静电势由Multiwfn[20]软件计 算。 2.5. 分子动力学模拟 为了进一步确认1,8-ANS 插层在锌铝,镁铝水滑 石层板的排布情况,采用分子动力学方法(MD)进行考 察。 水滑石层板结构模型以 Mg/Al 水滑石结构(空间 群R-3 m)为基础,分别构建 Mg/Al-LDHs 和Zn/Al- LDHs 6 × 6 × 1 的菱形超晶胞;模型的化学式为: 24 1248 12 MAlOH1,8-ANS, MMgZn 采用自行开发的LDHFF 力场[21],模拟采用NPT Copyright © 2013 Hanspub 201  M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 Copyright © 2013 Hanspub 202 佐证了 ONIOM 方法预测的1,8-ANS 在层板中的朝 向。但是层间 1,8-ANS分子构象各有不同,而且排列 并不整齐,而且插层到锌铝水滑石层板中的 1,8-ANS 比插层到镁铝水滑石层板中的略显紧凑。 系综;温度为298 K,压强0.1 MPa,分别使用 Andersen 法和 Berendsen法[22]控制;模拟时间为 100 ps,步长 为1 fs。结果显示,系统在模拟开始后 30 ps~40 ps 后 即达到结构和能量稳定状态,故在接下来的 60 ps 内, 每50 fs 记录一次动力学轨点(dynamic trajectories)。以 上模拟过程均在 Materials Studio 4.0 (Accelrys Inc., San Diego)软件包中的 Discover模块中运行。 3.2. 1,8-ANS与层板团簇单体的前线轨道 图7是优化出的镁铝(1)、锌铝(2)水滑石层板团簇 的结构。 3. 结果与讨论 根据优化出的镁铝、锌铝水滑石团簇几何结构, 可以明显看到,团簇中全部 H由于与电负性很强的氧 原子而显正电性,预计可以和1,8-ANS 具有较强吸电 子作用的磺酸基上的氧形成有较强相互作用。 3.1. 1,8-ANS层间取向的计算 3.1.1. ONIMO方法计算结果 使用 ONIOM 的方法得到 1,8-ANS在层间的取向 如图 8所示。 而且由表1可知,由于镁铝、锌铝水滑石的前线 轨道轨道能量与 1,8-ANS的前线轨道轨道能量相差较 大,故无论是 1,8-ANS的占据轨道与镁铝、锌铝水滑 石团簇的空轨道,还是 1,8-ANS 的空轨道与镁铝、锌 铝水滑石团簇的占据轨道之间都不会产生强的相互 作用,因而 1,8-ANS 与水滑石团簇之间不可能形成化 学键,而是主要是依靠弱相互作用力(包括可能出现的 氢键,取向力,诱导力,色散力等)来维持体系的稳定。 图中可以看出 ANS 由于磺酸基带较多电荷而导 致磺酸基靠近层板。镁铝,锌铝两种层板中结果是一 致的。 3.1.2. 分子动力学计算层间 1,8-ANS排布 分子动力学分析结果如图 6所示。 结果表明,1,8-ANS 在层板中磺酸基靠近层板, (a) (b) Figure 8. Molecular electronic potential (MEP) of Mg/ZnAl cluster and 1,8-ANS (a)/(b). For the MgAl/ZnAl cluster, blue points represent th e minima of MEP and red points represent the maxima; For 1,8-ANS, blue points represent the maxima of MEP and red points represent the minima. The regions in the elliptical in the LDHs clusters are close to 1,8-ANS. 1,8-ANS and Mg/ZnAl cluster show opposite charge. 图8. Mg/ZnAl水滑石团簇与 1,8-ANS 分子静电势(1)/(2)。Mg/ZnAl 团簇中蓝色表示静电势极小值点,红色为极大值点。1,8-ANS 中蓝色为 极大值点,浅红为极小值点。Mg/ZnAl 团簇中圈出部分为与 1,8-ANS 的磺酸基靠近的区域。1,8-ANS 与Mg/ZnAl团簇电荷异号 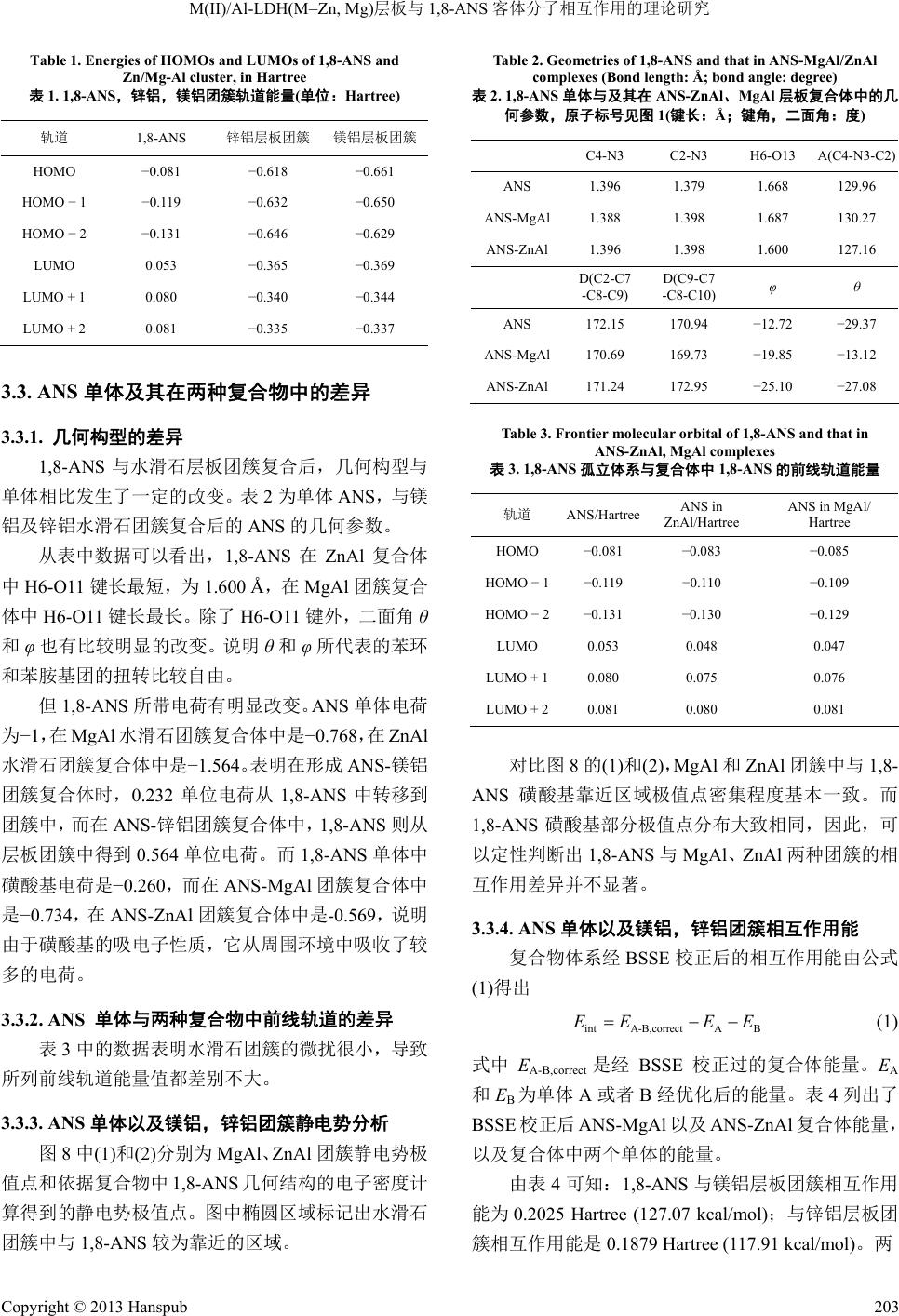 M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 Table 1. Energies of HOMOs and LUMOs of 1,8-ANS and Zn/Mg-Al cluster, in Hartree 表1. 1,8-ANS,锌铝,镁铝团簇轨道能量(单位:Hartree) 轨道 1,8-ANS 锌铝层板团簇 镁铝层板团簇 HOMO −0.081 −0.618 −0.661 HOMO − 1 −0.119 −0.632 −0.650 HOMO − 2 −0.131 −0.646 −0.629 LUMO 0.053 −0.365 −0.369 LUMO + 1 0.080 −0.340 −0.344 LUMO + 2 0.081 −0.335 −0.337 3.3. ANS单体及其在两种复合物中的差异 3.3.1. 几何构型的差异 1,8-ANS与水滑石层板团簇复合后,几何构型与 单体相比发生了一定的改变。表 2为单体 ANS,与镁 铝及锌铝水滑石团簇复合后的ANS 的几何参数。 从表中数据可以看出,1,8-ANS在ZnAl 复合体 中H6-O11 键长最短,为1.600 Å,在 MgAl 团簇复合 体中 H6-O11键长最长。除了 H6-O11键外,二面角 θ 和φ也有比较明显的改变。说明 θ和φ所代表的苯环 和苯胺基团的扭转比较自由。 但1,8-ANS 所带电荷有明显改变。ANS 单体电荷 为−1,在 MgAl 水滑石团簇复合体中是−0.768,在 ZnAl 水滑石团簇复合体中是−1.564。表明在形成 ANS-镁铝 团簇复合体时,0.232 单位电荷从 1,8-ANS 中转移到 团簇中,而在ANS-锌铝团簇复合体中,1,8-ANS 则从 层板团簇中得到 0.564 单位电荷。而1,8-ANS单体中 磺酸基电荷是−0.260,而在 ANS-MgAl团簇复合体中 是−0.734,在 ANS-ZnAl 团簇复合体中是-0.569,说 明 由于磺酸基的吸电子性质,它从周围环境中吸收了较 多的电荷。 3.3.2. ANS 单体与两种复合物中前线轨道的差异 表3中的数据表明水滑石团簇的微扰很小,导致 所列前线轨道能量值都差别不大。 3.3.3. ANS单体以及镁铝,锌铝团簇静电势分析 图8中(1)和(2)分别为 MgAl、ZnAl 团簇静电势极 值点和依据复合物中 1,8-ANS几何结构的电子密度计 算得到的静电势极值点。图中椭圆区域标记出水滑石 团簇中与1,8-ANS 较为靠近的区域。 Table 2. Geometries of 1,8-ANS and that in ANS-MgAl/ZnAl complexes (Bond length: Å; bond angle: degree) 表2. 1,8-ANS单体与及其在 ANS-ZnAl、MgAl 层板复合体中的几 何参数,原子标号见图 1(键长:Å;键角,二面角:度) C4-N3 C2-N3 H6-O13 A(C4-N3-C2) ANS 1.396 1.379 1.668 129.96 ANS-MgAl1.388 1.398 1.687 130.27 ANS-ZnAl 1.396 1.398 1.600 127.16 D(C2-C7 -C8-C9) D(C9-C7 -C8-C10) φ θ ANS 172.15 170.94 −12.72 −29.37 ANS-MgAl170.69 169.73 −19.85 −13.12 ANS-ZnAl 171.24 172.95 −25.10 −27.08 Table 3. Frontier molecular orbital of 1,8-ANS and that in ANS-ZnAl, MgAl complexes 表3. 1,8-ANS孤立体系与复合体中 1,8-ANS 的前线轨道能量 轨道 ANS/Hartree ANS in ZnAl/Hartree ANS in MgAl/ Hartree HOMO −0.081 −0.083 −0.085 HOMO − 1 −0.119 −0.110 −0.109 HOMO − 2 −0.131 −0.130 −0.129 LUMO 0.053 0.048 0.047 LUMO + 1 0.080 0.075 0.076 LUMO + 2 0.081 0.080 0.081 对比图 8的(1)和(2),MgAl 和ZnAl团簇中与 1,8- ANS 磺酸基靠近区域极值点密集程度基本一致。而 1,8-ANS磺酸基部分极值点分布大致相同,因此,可 以定性判断出1,8-ANS 与MgAl、ZnAl两种团簇的相 互作用差异并不显著。 3.3.4. ANS单体以及镁铝,锌铝团簇相互作用能 复合物体系经 BSSE 校正后的相互作用能由公式 (1)得出 intA-B,correct A B EE EE (1) 式中 EA-B,correct 是经 BSSE 校正过的复合体能量。EA 和EB为单体 A或者 B经优化后的能量。表 4列出了 BSSE 校正后 ANS-MgAl 以及ANS-ZnAl 复合体能量, 以及复合体中两个单体的能量。 由表 4可知:1,8-ANS 与镁铝层板团簇相互作用 能为 0.2025 Hartree (127.07 kcal/mol);与锌铝层板团 簇相互作用能是0.1879 Hartree (117.91 kcal/mol)。两 Copyright © 2013 Hanspub 203  M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 Table 4. Interaction between 1,8-ANS and MgAl or ZnAl cluster 表4. 1,8-ANS与镁铝,锌铝团簇能量及相互作用能(Hartree) 1,8-ANS 层板团簇 复合物 相互作用能 ANS-MgAl −1295.6347 −994.6375 −2290.4747 0.2025 ANS-ZnAl −1123.9736 −2419.7962 0.1879 者相互作用能差别为 9.17 kcal/mol。说明 1,8-ANS与 镁铝水滑石团簇相互作用能稍强于它与锌铝水滑石 团簇的相互作用。从总体能量上看,锌铝-ANS 水滑 石体系较镁铝-ANS体系更为稳定。 4. 结论 1) ONIOM方法和分子动力学计算表明 1,8-ANS 在水滑石层间的取向是磺酸基与层板接近; 2) 根据1,8-ANS的LUMO轨道能量与镁铝,锌 铝水滑石层板团簇的 HOMO轨道能量的对比,可知 1,8-ANS和水滑石层板团簇仅存在弱相互作用。 3) 通过静电势分析得知 1,8-ANS与锌铝、镁铝团 簇相互作用差别很小。根据基组重叠误差校正计算得 到1,8-ANS与层板团簇的相互作能表明,1,8-ANS 与 镁铝层板相互作用稍强于锌铝层板团簇相互作用。从 总体能量上看,锌铝-ANS 水滑石体系较镁铝-ANS 体 系更为稳定。本文从微观角度说明了层板与客体间的 相互作用,对构筑新型层状无机-有机超分子复合结构 体系提供了理论参考。 5. 致谢 感谢国家自然科学基金的资助。 参考文献 (References) [1] F. Cañaveras, R. Madueño, J. Sevilla, M. Blazquez and T. Pine- da. Role of the functionalization of the gold nanoparticle surface on the formation of bioconjugates with human serum albumin. The Journal of Physical Chemistry, C, 2012, 116(18): 10430- 10437. [2] A. M. Tan, M. T. Henzl. Evindence for a Ca2+-specific confor- mational change in avian thymic hormone, a high-affinity β-par- valbumin. Biochemistry, 2009, 48(18): 3936-3945. [3] M. Mehrabia, S. Ghobadi and R. Khodarahmi. Spectroscopic study on the interaction of celecoxib with human carbonic anhy- drase II: Thermodynamic characterization of the binding process, Journal of Photochemistry and Photobiology B, 2009, 97(2): 161-168. [4] F. Dinga, Y. Suna, J. X. Diao, X. N. Li, X. L. Yang, Y. Sun and L. Zhang. Features of the complex of food additive hesperidin to hemoglobin. Journal of Photochemistry and Photobiology B, 2012, 106(5): 53-60. [5] 许金钩, 王尊本. 荧光分析法[M]. 北京: 科学出版社, 2006: 65-69. [6] H. J. Buschmann, T. Wolff. Fluorescence of 1-anilinonaphtha- lene-8-sulfonate in solid macrocyclic environments. Journal of Photochemistry and Photobiology A, 1999, 121: 99-103. [7] 杜以波, D. G. Evans, 孙鹏, 段雪. 阴离子型层状柱材料研究 进展[J]. 化学通报, 2000, 5: 20-24. [8] M. F. Shao, J. B. Han, M. Wei, D. G. Evans and X. Duan. The synthesis of hierarchical Zn-Ti layered double hydroxide for efficient visible-light photocatalysis. Chemical Engineering Jour- nal, 2011, 168(1): 519-524. [9] Z. J. Huang, P. X. Wu, Y. H. Lu, X. R. Wang, N. W. Zhu and Z. Dang. Enhancement of photocatalytic degradation of dimethyl phthalate with nano-TiO2 immobilized onto hydrophobic layered double hydroxides: A mechanism study. Journal of Hazardous Materials, 2013, 15: 70-78. [10] G. Carja, L. Dartu, K, Okada and E. Fortunato. Nanoparticles of copper oxide on layered double hydroxides and the derived solid solutions as wide spectrum active nano-photocatalysts. Chemical Engineering Journal, 2013, 222(15): 60-66. [11] Z. Y. Sun, L. Jin, W. Y. Shi , M. Wei and X. Duan. Preparation of an anion dye intercalated into layered double hydroxides and its controllable luminescence properties. Chemical Engineering Jour- nal, 2010, 293-300. [12] H. Yan, M. Wei, J. Ma, F. Li, D. G. Evans and X. Duan. Theo- retical study on the structural properties and relative stability of M(II)-Al layered double hydroxides based on a cluster model. The Journal of Physical Chemistry A, 113(21): 6133-6147. [13] M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sie- ber and K. Morokuma. ONIOM: A multilayered integrated MO + MM method for geometry optimizations and single point en- ergy predictions. A test for diels-alder reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. The Journal of Physical Chemistry, 1996, 100(50): 19357-19363. [14] R. Allmann. The crystal structure of pyroaurite. Acta Crystal- lographica Section B, 1968, 24(7): 972-977. [15] S. F. Boys, F. Bernardi, Calculation of small molecular interact- tions by differences of separate total energies some procedures with reduced errors. Molecular Physics, 1970, 19(4): 553-566. [16] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dan- nenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Pittsburgh, 2003. [17] A. D. Beck. Density functional thermochemistry. IV. A new dy- namical correlation functional and implications for exact-ex- change mixing. Journal of Chemical Physics, 1996, 104(3): 1040- 1046. [18] J. A. Pople, R. K. Nesbet. Self-Consistent orbitals for radicals. Journal of Chemical Physics, 1954, 22(3): 571-572. [19] P. J. Hay, W. R. Wadt. Ab inito effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. Journal of Chemical Physics, 1985, 82(1): 270-283. [20] Lu T, Chen F W. Multiwfn: A multifunctional wavefunction ana- lyzer. Journal of Computational Chemistry, 2012, 33: 580-592 [21] S. T. Zhang, H. Yan, M. Wei, D. G. Evans and X. Duan. Valence force field for layered double hydroxide materials based on the Copyright © 2013 Hanspub 204  M(II)/Al-LDH(M=Zn, Mg)层板与1,8-ANS 客体分子相互作用的理论研究 Copyright © 2013 Hanspub 205 parameterization of octahedrally coordinated metal cations. The Journal of Physical Chemistry C, 2012, 116(5): 3421-3431. [22] A. K. Rappe, W. A. Goddard. Charge equilibration for molecular dynamics simulations. The Journal of Physical Chemistry, 1991, 95(8): 3358-3363. |