 Hans Journal of Chemical Engineering and Technology 化学工程与技术, 2013, 3, 169-174 http://dx.doi.org/10.12677/hjcet.2013.35030 Published Online September 2013 (http://www.hanspub.org/journal/hjcet.html) The Theoretical Study of Selective Oxidation of Styrene on Co3O4(112) Surface Haichuan Guo, Zuoyin Yang, Yaping Li* Beijing University of Chemical Technology, Beijing Email: *liyp@mail.buct.edu.cn Received: Jun. 25th, 2013; revised: Jul. 20th, 2013; accepted: Jul. 29th, 2013 Copyright © 2013 Haichuan Guo et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract: In this work, quantum mechanical method of density functional theory is introduced to study the reaction of styrene and TBHP on the Co3O4(112) surface. First, styrene reacts with an oxygen atom on the Co3O4 surface and de- sorbs with leaving an oxygen vacancy. Second, TBHP adsorbs on the crystal surface and fills the vacancy with one oxy- gen atom in itself. Keywords: Co3O4(112) Surface; Styrene; TBHP; Catalysis Reaction Mechanism; Density Functional Theory Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 郭海川,杨作银,李亚平* 北京化工大学,北京 Email: *liyp@mail.buct.edu.cn 收稿日期:2013 年6月25 日;修回日期:2013年7月20 日;录用日期:2013年7月29 日 摘 要:本文使用量子力学密度泛函方法对 Co3O4(112)晶面催化苯乙烯与 TBHP的反应进行了理论计算,探索 了苯乙烯分子在 Co3O4与晶面上 O2f位O原子相互作用生成环氧产物并脱附的过程,以及 TBHP 分子与晶面反 应,用自身的一个O原子填补 O2f 空位,生成叔丁醇最终脱附的过程。 关键词:Co3O4(112)晶面;苯乙烯;TBHP;催化;密度泛函理论 1. 引言 催化剂是现代化学工业不可或缺的重要组成部 分,是化工生产中高效利用原材料,节能减排,实现 可持续发展的重要手段。开发新型催化剂从而改进现 有生产方式始终是化学工作者不懈追求的目标。在工 业生产中,烃类的选择性氧化反应是一类重要的有机 反应,它是许多化工产品生产过程中的重要环节。其 中,烯烃的选择性氧化反应就有非常广泛的应用。目 前,在这类反应中,金属银是非常重要的催化剂。虽 然它有着很好的催化性能,但是作为一种贵金属,银 的使用无疑提高了生产成本。因此,为这类反应寻找 更加经济高效的催化剂就有着重要的现实意义。在新 型催化剂的开发领域,近年来,研究者们合成了许多 具有优良催化性能的Co3O4纳米颗粒[1-9],其中的一些 更是在催化烯烃的选择性氧化反应上有着出色的表 现,具备继续研究开发的价值。例如,Sun 等人[2]通 过水热合成等方法制备了多级Co 3 − xFexO4纳米阵列, 发现它催化苯乙烯氧化反应具有 92.2% 的转化率和 64.6%的苯甲醛选择性。又如 Askarinejad 等人[9]合成 *通讯作者。 Copyright © 2013 Hanspub 169  Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 的Co3O4纳米颗粒对叔丁基过氧化氢(TBHP)氧化苯 乙烯生成环氧化合物的反应有98 %的转化率,而块体 (微米级)Co3O4仅有14 %。这说明 Co3O4纳米颗粒高的 比表面积和暴露出的特殊晶面是催化活性的关键。 目前,对于 Co3O4这类金属氧化物催化剂的研究 大多数停留在实验室合成表征与性能测试阶段,对 Co3O4催化反应的理论研究也仅仅局限在 CO 氧化等 少数反应上,特别是对于烯烃在Co3O4纳米颗粒上选 择性氧化的机理的研究还是空白。用理论方法从更加 广泛的角度上分析催化反应的原理,不但有助于更加 深入地理解 Co3O4催化性能的本质,而且有助于指导 合成性能更加优良的Co3O4纳米颗粒,从而推动新型 催化剂的研究开发,有着十分重要的理论研究和实际 应用价值。因此,本文的目的就在于通过理论计算更 加深入地探索 Co3O4纳米颗粒催化苯乙烯选择性氧化 反应的机理,阐明Co3O4晶面结构和催化性质的关系, 并最终期待用理论成果指导实践。 本文使用量子力学计算软件VA S P研究了Co3O4 具备高催化活性的(112)晶面催化苯乙烯选择性氧化 反应的过程,以及 TBHP作用于反应后的晶面并且使 催化剂再生的过程。由于Co3O4与苯乙烯及TBHP 的 反应的机理尚不清楚,本文借鉴了 Co3O4催化氧化 CO 反应[10]以及 Mo 的环戊二烯基配合物催化TBHP 氧化 烯烃反应[11]的思路,将二者结合起来应用到本文的研 究当中,并通过理论计算加以验证和改进,进而通过 分析反应的过程和能量变化来探索晶面结构和反应 活性的关系。 2. 计算模型与方法 2.1. 计算模型 与(100)、(110)等常见晶面相比,Co3O4(112)晶面 结构十分复杂,表面形貌起伏相当大,特别是经过了 表面弛豫后,表面形态更加不规则。由于到目前为止 的研究尚未涉及(112)晶面表面形态构造,晶面计算模 型的选取就成了首要问题。为此,首先建立了四个不 同的晶面模型,如图1(a)、(b)、(c)、(d)所示。 这四个模型均为包含84个原子(Co 原子36个, O原子 48 个)的(112)晶面 p (1 × 2)模型,晶面面积为 11.56 Å × 14.16 Å,且在晶面上方设置了厚度为12.5 Å 的真空层。在计算时固定50%左右的原子,表层原子 Figure 1. Calculation models of Co3O4(112); (a) model a; (b) model b; (c) model c; (d) model d 图1. Co3O4(112)晶面计算模型;(a) 模型 a;(b) 模型 b; (c) 模型 c;(d) 模型 d 弛豫。 由于缺少 Co3O4(11 2)晶面表层形貌研究的数据, 所以需要选择一个合理的计算模型。为此,计算了这 四个模型弛豫后的总能量及单位面积能量并进行比 较,结果如表1所示. 从数据可以看出,模型b计算得到的能量最高。 众所周知,晶面的催化性能与表面能量是紧密联系 的。一般来说,晶面能量越高,催化活性也就越高。 根据这一规律,以及晶面弛豫后的变形程度等因素进 行综合考虑,本文选择了模型b作为计算模型进行接 下来的研究。 从图 1(b)中可以看出,Co3O4(112)晶面模型 b的 表面 O原子根据结构可分为四类,分别称为Oa、Ob、 O2f 和O3f。O2f和O3f 分别指与两个 Co 原子成键的 O 原子和与三个Co 原子成键的 O原子。根据计算结果, Oa与Ob位O原子离开晶面形成空位后,晶面结构变 化过大,不利于晶面再生,因此暂不讨论。本文中选 择了有着较弱配位场,与相邻原子结合相对较弱的 O2f 位O原子作为研究对象。 2.2. 计算方法 在计算方法上,使用VASP 软件中的密度泛函 (DFT)方法进行结构优化和能量计算。对于电子交换 Copyright © 2013 Hanspub 170  Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 Table 1. Energy of calculation models of Co3O4(112) 表1. Co3O4(112)晶面计算模型能量 模型 总能/eV 单位面积能量/eV·Å−2 a −466.305 −2.847 b −457.342 −2.792 c −462.001 −2.821 d −465.492 −2.842 关联势采用了自旋极化(spin-polarized)的广义梯度近 似(GGA)中的 Perdew-Burke-Ernzerhof (PBE)泛函进行 计算。用投影缀加波 PAW (Projector Augmented Wave) 来描述原子核与电子之间的相互作用。截断能设置为 400 eV。由于计算模型体积较大且有相当厚的真空层, 因此对于布里渊区的设置,采用了以原点为中心的1 × 1 × 1的Monkhorst-Pack 网格 k点空间取样。由于 Co3O4有较强的电子相关作用,在计算时使用了 DFT + U方法并将 U值设置为 3.5。在计算精度上,由于 原子数量过多(包括有机分子共 100 个),计算体系过 于庞大,因此,在实际计算中将优化过程分两步进行: 首先,使用1 × 10−4 eV 的电子步收敛标准,将结构优 化到单个原子受力小于0.2 eV/Å;在此基础上,将电 子步收敛标准提高到1 × 10−5 eV,再最终将体系优化 到单个原子受力小于0.05 eV/Å。 3. 结果与讨论 3.1. 苯乙烯与 Co3O4(112)晶面的反应 首先将苯乙烯分子置于(112)晶面模型的O2f 位上 方3.5 Å 左右,优化结构,得到最初的吸附态 S1,如 图2(a)所示。 从图中可以看出,苯乙烯分子整体并不与晶面平 行,而是呈倾斜状态,乙烯基更加靠近晶面,表明(112) 晶面的表面势场并不均匀,这与晶面的高低起伏是有 关系的。此时,C1-O 距离为 3.855 Å,C2-O 距离为 3.927 Å,没有有效成键,苯乙烯分子处于物理吸附状 态。 在此基础上,苯乙烯分子继续靠近晶面,与晶面 上的 O2f 反应,生成环状结构并吸附于晶面之上(图2 b,吸附态 S2),C1-O 键长1.464 Å。此时 O2f 位O原 子受到 C1、C2 的吸引,稍稍高于原位,O-Co1、O-Co2 键长从初始的1.915 Å、1.726 Å 拉长到 2.294 Å、2.049 C1、C2 为苯乙烯中乙烯基 C原子,C3 为苯环上与乙烯基相 连的 C原子,O为晶面上与乙烯基距离最近的 O2f 位O原子, Co1、Co2 分别为与 O相连的次表层和表层 Co 原子 Figure 2. Adsorption state S1; (a) adsorption state S2; (b) styrene on Co3O4(112) 图2. 苯乙烯与 Co3O4(112)晶面反应过程中的结构;(a) 吸附态 S1; (b) 吸附态 S2 Å。同时,C2-C3 键长也因为碳碳双键被破坏、与苯 环的共轭程度减弱而拉长,由最初的 1.468 Å 变为 1.476 Å。此时,与物理吸附态相比,能量下降了275.41 kJ·mol−1,说明(112)晶面的O2f 有较高的氧化能力。 从Co3O4(112)晶面的电荷密度图(图3)中可以看 出,苯乙烯分子与晶面的O原子成键前,乙烯基双键 与苯环形成一个大的共轭体系;而在成键后,共轭体 系被破坏,乙烯基与O原子周围的电荷相向移动,两 者之间电荷密度增加,形成三元环结构。与此同时, (112)晶面上许多原子的电荷密度都发生了改变,这也 说明了(112)晶面的复杂性。 接下来,环氧化的苯乙烯分子逐步脱附,离开晶 体表面。首先,产物分子由最初的吸附位置上升, O-Co1 距离拉大到 4.288 Å,此时可以认为O原子已 与晶面次表层的 Co1 之间已没有相互作用,O2f 已脱 离晶格原有位置,吸附在表层之上(图4a,吸附态 S3), O-Co2键长为 2.179 Å。该过程是一个能量上升的过 程,体系能量增加30.95 kJ·mol−1。随后,产物分子继 续上升远离晶面,O-Co2键长增加到 4.829 Å,体系 能量再次上升71.77 kJ·mol−1,最终完成化学脱附过 程,留下一个 O2f 空位(图4b,吸附态 S4)。从总体来 说,苯乙烯分子在(112)晶面上的氧化反应放出热量 172.69 kJ·mol−1,也说明了(112)晶面的 O2f 的高活性。 反应过程能量变化情况见图5。反应过程中各状态键 长数据见表2。 3.2. TBH P与Co3O4(112)晶面的反应 在上一步反应中,苯乙烯分子与晶面上的O原子 Copyright © 2013 Hanspub 171 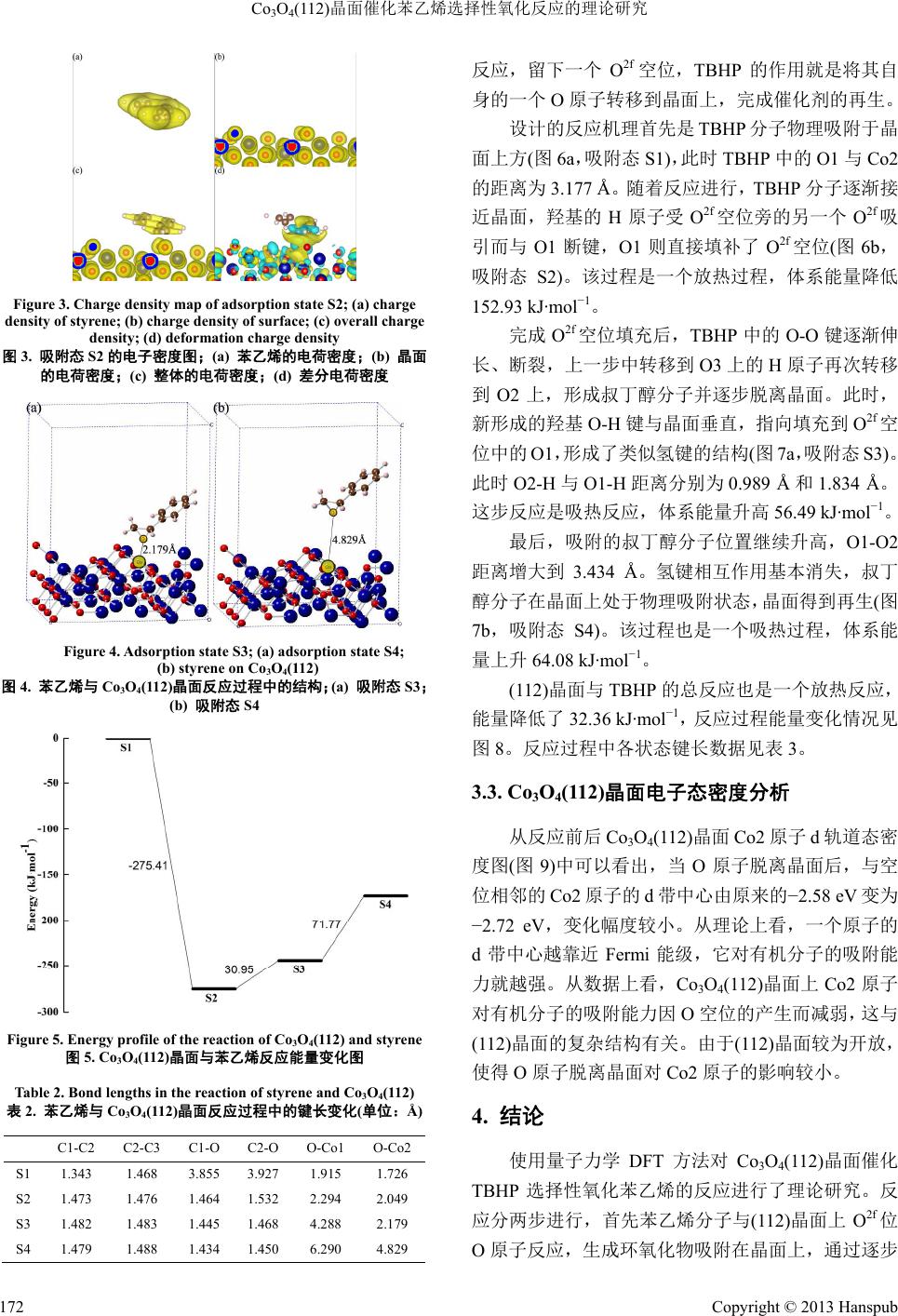 Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 Figure 3. Charge density map of adsorption state S2; (a) charge density of styrene; (b) charge density of surface; (c) overall charge density; (d) deformation charge density 图3. 吸附态 S2 的电子密度图;(a) 苯乙烯的电荷密度;(b) 晶面 的电荷密度;(c) 整体的电荷密度;(d) 差分电荷密度 Figure 4. Adsorption state S3; (a) adsorption state S4; (b) styrene on Co3O4(112) 图4. 苯乙烯与 Co3O4(112)晶面反应过程中的结构;(a) 吸附态 S3; (b) 吸附态 S4 Figure 5. Energy profile of the reaction of Co3O4(112) and styrene 图5. Co3O4(112)晶面与苯乙烯反应能量变化图 Table 2. Bond lengths in the reaction of styrene and Co3O4(112) 表2. 苯乙烯与 Co3O4(112)晶面反应过程中的键长变化(单位:Å) C1-C2 C2-C3 C1-O C2-O O-Co1 O-Co2 S1 1.343 1.468 3.855 3.927 1.915 1.726 S2 1.473 1.476 1.464 1.532 2.294 2.049 S3 1.482 1.483 1.445 1.468 4.288 2.179 S4 1.479 1.488 1.434 1.450 6.290 4.829 反应,留下一个O2f 空位,TBHP 的作用就是将其自 身的一个O原子转移到晶面上,完成催化剂的再生。 设计的反应机理首先是 TBHP 分子物理吸附于晶 面上方(图6a,吸附态S1),此时 TBHP 中的O1 与Co2 的距离为 3.177 Å。随着反应进行,TBHP 分子逐渐接 近晶面,羟基的 H原子受O2f 空位旁的另一个 O2f吸 引而与 O1断键,O1 则直接填补了 O2f空位(图6b, 吸附态 S2)。该过程是一个放热过程,体系能量降低 152.93 kJ·mol−1。 完成 O2f空位填充后,TBHP 中的 O-O 键逐渐伸 长、断裂,上一步中转移到O3 上的 H原子再次转移 到O2上,形成叔丁醇分子并逐步脱离晶面。此时, 新形成的羟基O-H 键与晶面垂直,指向填充到O2f 空 位中的 O1,形成了类似氢键的结构(图7a,吸附态S3)。 此时 O2-H 与O1-H 距离分别为 0.989 Å和1.834 Å。 这步反应是吸热反应,体系能量升高 56.49 kJ·mol−1。 最后,吸附的叔丁醇分子位置继续升高,O1-O2 距离增大到 3.434 Å。氢键相互作用基本消失,叔丁 醇分子在晶面上处于物理吸附状态,晶面得到再生(图 7b,吸附态 S4)。该过程也是一个吸热过程,体系能 量上升 64.08 kJ·mol−1。 (112)晶面与 TBHP 的总反应也是一个放热反应, 能量降低了32.36 kJ·mol−1,反应过程能量变化情况见 图8。反应过程中各状态键长数据见表3。 3.3. Co3O4(112)晶面电子态密度分析 从反应前后 Co3O4(112)晶面 Co2 原子 d轨道态密 度图(图9)中可以看出,当 O原子脱离晶面后,与空 位相邻的 Co2 原子的 d带中心由原来的−2.58 eV变为 −2.72 eV,变化幅度较小。从理论上看,一个原子的 d带中心越靠近 Fermi 能级,它对有机分子的吸附能 力就越强。从数据上看,Co3O4(112)晶面上Co2 原子 对有机分子的吸附能力因O空位的产生而减弱,这与 (112)晶面的复杂结构有关。由于(112)晶面较为开放, 使得 O原子脱离晶面对Co2 原子的影响较小。 4. 结论 使用量子力学 DFT方法对 Co3O4(112)晶面催化 TBHP选择性氧化苯乙烯的反应进行了理论研究。反 应分两步进行,首先苯乙烯分子与(112)晶面上O2f 位 O原子反应,生成环氧化物吸附在晶面上,通过逐步 Copyright © 2013 Hanspub 172 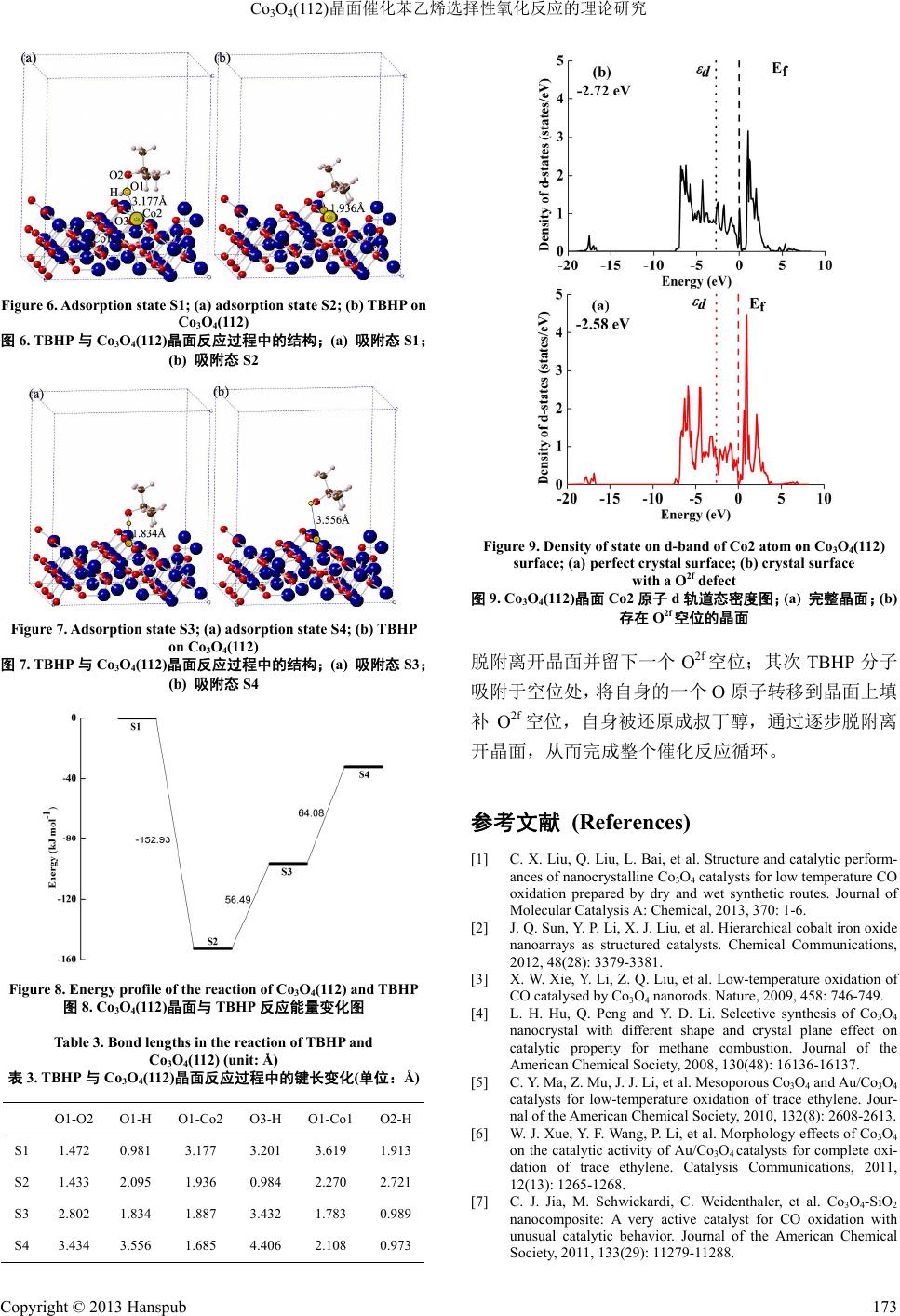 Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 Figure 6. Adsorption state S1; (a) adsorption state S2; (b) TBHP on Co3O4(112) 图6. T BH P与Co3O4(112)晶面反应过程中的结构;(a) 吸附态 S1; (b) 吸附态 S2 Figure 7. Adsorption state S3; (a) adsorption state S4; (b) TBHP on Co3O4(112) 图7. T BH P与Co3O4(112)晶面反应过程中的结构;(a) 吸附态 S3; (b) 吸附态 S4 Figure 8. Energy profile of the reaction of Co3O4(112) and TBHP 图8. Co3O4(112)晶面与 TBHP 反应能量变化图 Table 3. Bond lengths in the reaction of TBHP and Co3O4(112) (unit: Å) 表3. T BH P与Co3O4(112)晶面反应过程中的键长变化(单位:Å) O1-O2 O1-H O1-Co2 O3-H O1-Co1 O2-H S1 1.472 0.981 3.177 3.201 3.619 1.913 S2 1.433 2.095 1.936 0.984 2.270 2.721 S3 2.802 1.834 1.887 3.432 1.783 0.989 S4 3.434 3.556 1.685 4.406 2.108 0.973 Figure 9. Density of state on d-band of Co2 atom on Co3O4(112) surface; (a) perfect crystal surface; (b) crystal surface with a O2f defect 图9. Co3O4(112)晶面 Co2 原子 d轨道态密度图;(a) 完整晶面;(b) 存在 O2f 空位的晶面 脱附离开晶面并留下一个 O2f 空位;其次 TBHP 分子 吸附于空位处,将自身的一个O原子转移到晶面上填 补O2f 空位,自身被还原成叔丁醇,通过逐步脱附离 开晶面,从而完成整个催化反应循环。 参考文献 (References) [1] C. X. Liu, Q. Liu, L. Bai, et al. Structure and catalytic perform- ances of nanocrystalline Co3O4 catalysts for low temperature CO oxidation prepared by dry and wet synthetic routes. Journal of Molecular Catalysis A: Chemical, 2013, 370: 1-6. [2] J. Q. Sun, Y. P. Li, X. J. Liu, et al. Hierarchical cobalt iron oxide nanoarrays as structured catalysts. Chemical Communications, 2012, 48(28): 3379-3381. [3] X. W. Xie, Y. Li, Z. Q. Liu, et al. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature, 2009, 458: 746-749. [4] L. H. Hu, Q. Peng and Y. D. Li. Selective synthesis of Co3O4 nanocrystal with different shape and crystal plane effect on catalytic property for methane combustion. Journal of the American Chemical Society, 2008, 130(48): 16136-16137. [5] C. Y. Ma, Z. Mu, J. J. Li, et al. Mesoporous Co3O4 and Au/Co3O4 catalysts for low-temperature oxidation of trace ethylene. Jour- nal of the American Chemical Society, 2010, 132(8): 2608-2613. [6] W. J. Xue, Y. F. Wang, P. Li, et al. Morphology effects of Co3O4 on the catalytic activity of Au/Co3O4 catalysts for complete oxi- dation of trace ethylene. Catalysis Communications, 2011, 12(13): 1265-1268. [7] C. J. Jia, M. Schwickardi, C. Weidenthaler, et al. Co3O4-SiO2 nanocomposite: A very active catalyst for CO oxidation with unusual catalytic behavior. Journal of the American Chemical Society, 2011, 133(29): 11279-11288. Copyright © 2013 Hanspub 173  Co3O4(112)晶面催化苯乙烯选择性氧化反应的理论研究 Copyright © 2013 Hanspub 174 [8] X. W. Xie, W. J. Shen. Morphology control of cobalt oxide nanocrystals for promoting their catalytic performance. Nano- scale, 2009, 1: 50-60. [9] A. Askarinejada, M. Bagherzadehb and A. Morsalia. Catalytic performance of Mn3O4 and Co3O4 nanocrystals prepared by sonochemical method in epoxidation of styrene and cyclooctene. Applied Surface Science, 2010, 256(22): 6678-6682. [10] D. E. Jiang, S. Dai. The role of low-coordinate oxygen on Co3O4(110) in catalytic CO oxidation. Physical Chemistry Chemical Physics, 2011, 13(3): 978-984. [11] P. J. Costa, M. J. Calhorda and F. E. Kühn. Olefin epoxidation catalyzed by η5-cyclopentadienyl molybdenum compounds: A computational study. Organometallics, 2010, 29(2): 303-311. |