Applied Physics
Vol.06 No.12(2016), Article ID:19266,7
pages
10.12677/APP.2016.612035
First-Principles Study of Structural and Electronic Properties of Perovskite-Type CH3NH3PbI3
Hongfei Liu1, Zhongwei Zhang2
1School of Science, Tianjin Chengjian University, Tianjin
2School of Computer and Information Engineering, Tianjin Chengjian University, Tianjin

Received: Nov. 28th, 2016; accepted: Dec. 13th, 2016; published: Dec. 16th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Using first-principle calculations based on density functional theory (DFT), we have studied the structural and electronic properties of perovskite-type CH3NH3PbI3. The structural properties cal- culated by DFT with the optB86b + vdWDF exchange-correlation functional are in good agreement with experimental results. The band structures analysis shows that perovskite-type CH3NH3PbI3 is a direct band gap material. In addition, the calculated band gap, ~1.68 eV, is close to experimental results. The analysis of partial density of states shows that the top of valence band is mainly composed of I 5p states. And, the main components of the conduction bands bottom are Pb 6p states. In contrast, the organic CH3NH3 makes little contribution to the top valence and bottom conduction bands. Thus, consideration of the vdW interactions is important for theoretical studies of perovskite-type CH3NH3PbI3 material.
Keywords:First-Principles, CH3NH3PbI3, Electronic Properties

钙钛矿CH3NH3PbI3结构和电子性质的 第一性原理研究
刘洪飞1,张众维2
1天津城建大学理学院,天津
2天津城建大学计算机与信息工程学院,天津

收稿日期:2016年11月28日;录用日期:2016年12月13日;发布日期:2016年12月16日

摘 要
我们采用基于密度泛函理论的第一性原理方法研究了钙钛矿CH3NH3PbI3材料的结构和电子性质。计算表明optB86b + vdWDF交换关联泛函给出的晶格结构和实验结果符合的很好。同时,能带结构分析表明材料是直接带隙材料,带隙宽度为1.68 eV与实验结果相符。态密度分析表明,价带顶和导带底分别是由I 5p轨道和Pb 6p轨道组成,而有机分子CH3NH3对于价带顶和导带底的贡献较小。这些结果说明,对于钙钛矿CH3NH3PbI3类材料,包含范德瓦尔斯作用的vdW-DF泛函能够更好的描述体系的物理性质。
关键词 :第一性原理,CH3NH3PbI3,电子性质

1. 引言
太阳能电池能够将太阳能直接转化为电能,可以为人类社会发展提供取之不尽用之不竭的清洁能源。目前,单晶硅基太阳电池技术已经比较成熟,但单晶硅基太阳电池存在生产成本高、生产过程能量消耗大、环境污染严重、成本回收时间长等问题。因此,新兴的太阳能电池技术不断涌现,包括染料敏化太阳电池 [1] [2] 、有机太阳电池和量子点太阳电池等。
2009年,日本Miyasaka等人在研究染料敏化太阳电池的过程中,首次使用具有钙钛矿结构的有机金属卤化物CH3NH3PbBr3和CH3NH3PbI3作为敏化剂 [3] ,拉开了钙钛矿太阳电池研究的序幕。在随后短短的几年时间内,钙钛矿太阳能电池技术取得了突飞猛进的进展 [4] [5] [6] [7] ,能量转换效率已经超过了染料敏化太阳电池、有机太阳电池和量子点太阳电池。2011年,Im等人将实验方案进行了改进与优化,制备的CH3NH3PbI3量子点电池的效率达到了6.54% [8] 。但是由于部分金属卤化物在液态电解质中发生溶解,很大程度上降低了电池的稳定性与使用寿命。2012年,Kim等人将Spiro-OMeTAD作为有机空穴传输材料应用到钙钛矿电池中,使钙钛矿电池的稳定性和工艺重复性大大提高 [9] 。此后,Burschka [10] 、Liu [11] 、Zhou [12] 等人采用采用不同的方法改进材料的性质,制备的钙钛矿太阳能电池的了转化效率超过15%,最高转化效率可以达到19.3%。钙钛矿铅卤化合物之所以能够高效的吸收太阳光,主要是因为其光学带隙宽度与太阳光谱的可见光区匹配较好。
随着研究范围的扩展,钙钛矿太阳能电池的结构也在不断发展变化,从最初使用液态电解质敏化太阳能电池的结构,发展到使用固态电解质敏化太阳能电池的结构,到逐步转化到平面异质节p-i-n结电池结构。目前,对于钙钛矿太阳电池中电荷输运机理还没有完全理解清楚,影响电池性能的关键因素,比如能带结构调控,也还需要进一步研究 [13] [14] [15] 。因此,本文采用基于密度泛函理论的第一性原理方法,研究了钙钛矿结构CH3NH3PbI3的结构和电子性质,分析了体系中原子之间的成键机理,以期对实验有一定的指导作用。
2. 计算方法和模型
本文所有的计算都是在基于密度泛函理论赝势平面波的VASP程序中进行的 [16] [17] [18] [19] 。电子与离子之间的相互作用通过PAW方法来描述,采用广义梯度近似(PBE)描述电子与电子之间的交换关联项。计算中所涉及到原子的价电子组态选择为:Pb:5d106s26p2,I:5s25p5,C:2s22p2,N:2s22p3,H:1s1。平面波的截断能量为500 eV,并采用以Γ为中心的5 × 5 × 5的格点对第一布里渊区的进行积分。自洽计算的收敛精度为10−5 eV∙atom−1。对于单胞形状和体积以及内部坐标进行优化,收敛标准为原子受力小于0.05 eV/Å。考虑到在包含着弱相互作用的材料中,比如有机无机杂化材料,范德瓦尔斯相互作用对于体系结构的影响也很重要 [20] 。因此,我们采用了optB86b + vdWDF的交换关联泛函形式来描述体系当中的范德瓦尔斯相互作用 [21] [22] 。
CH3NH3PbI3在不同温度下具有不同的结构:在低温条件下,材料为正交晶系结构(空间群:Pnma);当温度大于161.4 K,材料转变为四方晶系结构(空间群:I4/mcm);当温度高于330.4 K,材料转变为对称性更高的立方相结构(空间群:Pm-3m)。由于只有在低温条件下,材料中有机分子的取向才会趋向于有规律的排列 [23] ,并且从实验上确定C和N原子的位置,因而本文的计算采用的是低温正交结构的CH3NH3PbI3,计算所采用的晶胞如图1所示。
3. 钙钛矿CH3NH3PbI3的结构和电子性质
表1给出了优化计算后得到的CH3NH3PbI3体系的晶格常数,实验上以及其他计算给出的结果也一并给出。从表中可以看出,我们采用采用非局域vdW-DF方法计算得到的晶格常数与实验值符合的很好,并且优于其他计算得到的结果。这说明对于有机无机杂化类的化合物,引入包含范德瓦尔斯作用的vdW-DF方法对于更好的描述体系的晶体结构是十分必要的。
钙钛矿CH3NH3PbI3的总态密度以及CH3NH3、Pb和I的分波态密度分布如图2所示。与一般第一性原理计算低估材料带隙的结果不同,我们计算得到CH3NH3PbI3的带隙宽度为1.68 eV,与实验上得到的结果相一致 [25] [26] 。这主要是由于Pb元素较强的相对论效应与典型GGA计算低估带隙的误差相抵消的结果,类似的结果在其它含Pb的材料中也有报道 [27] [28] ,但这并不影响我们分析钙钛矿CH3NH3PbI3的电子结构性质。
通过比较CH3NH3、Pb和I的分波态密度分布,我们可以得到体系原子之间的成键机理。对于钙钛矿结构的ABX3类化合物来说,体系原子之间的半径比值 越接近于1,体系的结构
越接近于1,体系的结构
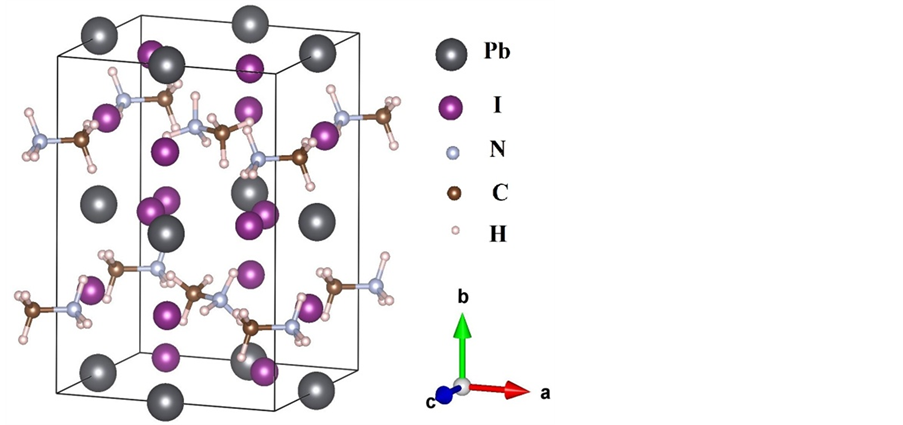
Figure 1. Crystal structure of perovskite-type CH3NH3PbI3
图1. 钙钛矿CH3NH3PbI3的结构图

Table 1. Calculated lattice constant of perovskite-type CH3NH3PbI3, as well as experimental value and other calculated results.
表1. 第一性原理计算得到的钙钛矿CH3NH3PbI3晶格常数。作为比较,实验结果和其他计算值也分别列出
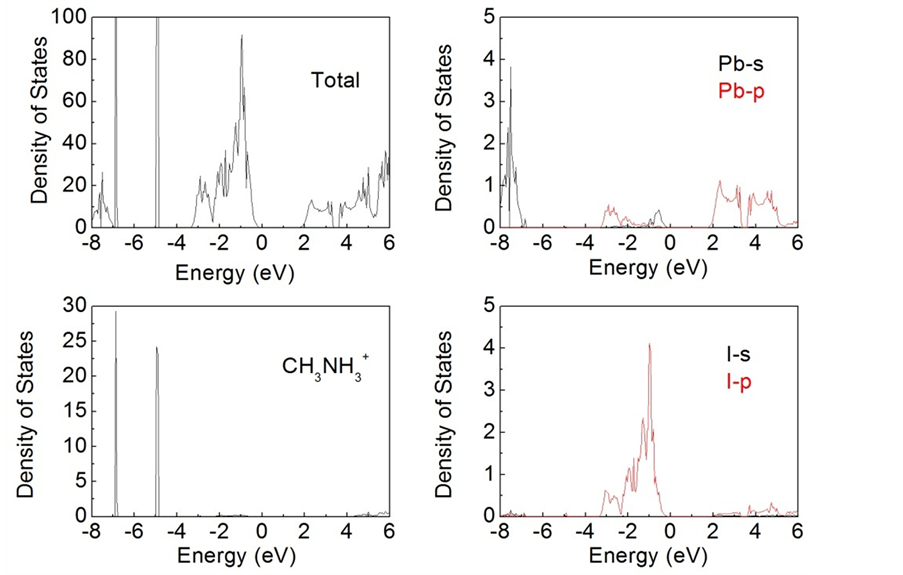
Figure 2. Total density of states of perovskite-type CH3NH3PbI3 and partial density of states of CH3NH3、Pb and I. The energy zero represents the fermi level
图2. 钙钛矿CH3NH3PbI3的总态密度以及CH3NH3、Pb和I的分波态密度分布,费米能级所在的位置设置为零
越稳定。为了满足要求,体系中A离子的半径要远大于B离子的半径 [29] 。对于CH3NH3PbI3来说,处于B位的Pb离子半径已经很大,很难找到满足条件的单元素A,而有机分子团CH3NH3的尺寸能够满足上面的条件,但其对于导带底和价带顶附近态密度的贡献则可以忽略不计。从图2可以看出,钙钛矿CH3NH3PbI3价带顶的态密度主要来自于I 5p轨道与Pb 6s轨道的贡献,而导带底的态密度主要来自于Pb 6p轨道的贡献。分波态密度分析表明,该材料吸收光子之后,电子将从价带顶的I 5p轨道跃迁到导带底未被占据的Pb 6p轨道。此外,Pb 6s轨道主要集中在−8.2 eV~−6.8 eV的导带深处,对于材料光吸收性质的影响较小。而C和N的2p轨道以及H的1s轨道主要集中在−6.8 eV和−4.8 eV附近的窄带,与Pb和I的s、p轨道重叠较小,表明有机分子CH3NH3和Pb、I之间不存在明显的共价相互作用。
钙钛矿CH3NH3PbI3体系的能带结构如图3所示。从图中可以看出,该材料属于直接带隙晶体(直接带隙宽度为1.68 eV),价带顶和导带底位于布里渊区的中心点Γ,最小的非直接带隙对应于Γ点到Y点(间接带隙宽度为1.92 eV)。总体上,CH3NH3PbI3的能带可以分为三部分:由C 2p、N 2p和H 1s轨道组成

Figure 3. Band structure of the orthorhombic phase of the CH3NH3PbI3. The path through the high symmetry points of orthorhombic lattice: Γ = (0,0,0), Z = (0,0,1/2), T = (0,1/2,1/2), Y = (0,1/2,0), S = (1/2,1/2,0), X = (1/2,0,0), U = (1/2,0,1/2), R = (1/2,1/2,1/2)
图3. 钙钛矿CH3NH3PbI3的能带结构。能带中包含的布里渊区高对称性点为:Γ = (0,0,0), Z = (0,0,1/2), T = (0,1/2,1/2), Y = (0,1/2,0), S = (1/2,1/2,0), X = (1/2,0,0), U = (1/2,0,1/2), R = (1/2,1/2,1/2)
的−4.8 eV附近的窄带,主要由I 5p轨道组成的价带顶以及主要由Pb 6p轨道组成的导带底。从图中可以看出,有机分子CH3NH3的能带是窄平状的,表明有机分子附近的电子是局域电子。而价带顶和导带底附近的能带展宽比较大,表明该区域的能态具有非局域的特性。尤其导带底的能带展宽较大,表明材料吸收光子使电子从价带跃迁到导带之后具有较高的迁移速度,这与实验上的结论相一致 [30] 。
4. 结论
我们采用基于密度泛函理论的第一性原理方法研究了钙钛矿CH3NH3PbI3材料的晶格结构和电子性质。计算结果表明,采用optB86b + vdWDF交换关联泛函计算得到的晶格结构与实验符合的很好,且优于未采用vdW泛函的其它计算结果。能带结构分析表明材料是直接带隙材料,价带顶和导带底位于布里渊区中心点Γ,带隙宽度为1.68 eV。态密度分析表明,价带顶和导带底分别是由I 5p轨道和Pb 6p轨道组成,而有机分子CH3NH3对于价带顶和导带底的贡献较小。这些结果说明,对于包含弱相互作用的有机无机杂化钙钛矿CH3NH3PbI3材料,采用引入范德瓦尔斯作用的vdW-DF泛函的第一性原理计算能够更好的描述体系的物理性质。
基金项目
本论文感谢国家自然科学基金(批准号:11547123)的资助。感谢天津城建大学科研启动项目(05140704, 05147001)的资助。
文章引用
刘洪飞,张众维. 钙钛矿CH3NH3PbI3结构和电子性质的第一性原理研究
First-Principles Study of Structural and Electronic Properties of Perovskite-Type CH3NH3PbI3[J]. 应用物理, 2016, 06(12): 281-287. http://dx.doi.org/10.12677/APP.2016.612035
参考文献 (References)
- 1. Tan, S., Zhai, J., Wan, M., Meng, Q., Li, Y., Jiang, L. and Zhu, D. (2004) Influence of Small Molecules in Conducting Polyaniline on the Photovoltaic Properties of Solid-State Dye-Sensitized Solar Cells. The Journal of Physical Chemistry B, 108, 18693-18697. https://doi.org/10.1021/jp046574y
- 2. Cervini, R., Cheng, Y. and Simon, G. (2004) Solid-State Ru-Dye Solar Cells Using Polypyr Role as a Hole Conductor. Journal of Physics D: Applied Physics, 37, 13. https://doi.org/10.1088/0022-3727/37/1/004
- 3. Kojima, A., Teshima, K., Shirai, Y. and Miyasaka, T. (2009) Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. Journal of the American Chemical Society, 131, 6050-6051. https://doi.org/10.1021/ja809598r
- 4. Lee, M.M., Teuscher, J., Miyasaka, T., Murakami, T.N. and Snaith, H.J. (2012) Efficient Hybrid Solar Cells Based on Meso-Superstructured Organometal Halide Perovskites. Science, 338, 643-647. https://doi.org/10.1126/science.1228604
- 5. Heo, J.H., Im, S.H., Noh, J.H., Mandal, T.N., Lim, C.S., Chang, J.A., Lee, Y.H., Kim, H.J., Sarkar, A. and Gratzel, M. (2013) Efficient Inorganic-Organic Hybrid Heterojunction Solar Cells Containing Perovskite Compound and Polymeric Hole Conductors. Nature Photonics, 7, 486-491. https://doi.org/10.1038/nphoton.2013.80
- 6. Noh, J.H., Im, S.H., Heo, J.H., Mandal, T.N. and Seok, S.I. (2013) Chemical Management for Colorful, Efficient, and Stable Inorganic-Organic Hybrid Nanostructured Solar Cells. Nano Letters, 13, 1764-1769. https://doi.org/10.1021/nl400349b
- 7. Stoumpos, C.C., Malliakas, C.D. and Kanatzidis, M.G. (2013) Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties. Inorganic Chemistry, 52, 9019-9038. https://doi.org/10.1021/ic401215x
- 8. Im, J.H., Lee, C.R., Lee, J.W., Park, S.W. and Park, N.G. (2011) 6.5% Efficient Perovskite Quantum-Dot-Sensitized Solar Cell. Nanoscale, 3, 4088-4093. https://doi.org/10.1039/c1nr10867k
- 9. Kim, H.S., Lee, C.R., Im, J.H., Lee, K.B., Moehl, T., Marchioro, A., Moon, S.J., Baker, R.H., Yum, J.H., Moser, J.E., Grätzel, M. and Park, N.G. (2012) Lead Iodide Perovskite Sensitized All-Aolid-Statesubmicron Thin Film Mesoscopic Solar Cell with Efficiency Exceeding 9%. Scientific Reports, 2, 591. https://doi.org/10.1038/srep00591
- 10. Burschka, J., Pellet, N., Moon, S.J., Baker, R.H., Gao, P., Nazeeruddin, M.K. and Grätzel, M. (2013) Sequential Deposition as a Route to High-Performance Perovskite-Sensitized Solar Cells. Nature, 499, 316-319. https://doi.org/10.1038/nature12340
- 11. Liu, M.Z., Johnston, M.B. and Snaith, H.J. (2013) Efficient Planar Heterojunction Perovskite Solar Cells by vaPour Deposition. Nature, 501, 395-398. https://doi.org/10.1038/nature12509
- 12. Zhou, H.P., Chen, Q., Li, G., Luo, S., Song, T.B., Duan, H.S., Hong, Z.R., You, J.B., Liu, Y.S. and Yang, Y. (2014) Interface Engineering of Highly Efficient Perovskite Solar Cells. Science, 345, 542-546. https://doi.org/10.1126/science.1254050
- 13. Kim, H.S., Im, S.H. and Park, N.G. (2014) Organolead Halide Perovskite: New Horizons in Solar Cell Research. The Journal of Physical Chemistry C, 118, 5615-5625. https://doi.org/10.1021/jp409025w
- 14. Shi, J.J., Dong, J., Lv, S.T., Xu, Y.Z., Zhu, L.F., Xiao, J.Y., Xu, X., Wu, H.J., Li, D.M., Luo, Y.H. and Meng, Q.B. (2014) Hole-Conductor-Free Perovskite Organic Lead Iodide Heterojunction Thin-Film Solar Cells: High Efficiency and Junction Property. Applied Physics Letters, 104, Article ID: 063901. https://doi.org/10.1063/1.4864638
- 15. Green, M.A., Baillie, A.H. and Snaith, H.J. (2014) Perovskite Solar Cells with a Planar Heterojunction Structure Prepared Using Room-Temperature Solution Processing Techniques. Nature Photonics, 8, 133-138.
- 16. Kresse, G. and Hafner, J. (1993) Ab Initio Molecular Dynamics for Liquid Metals. Physical Review B, 47, 558-561. https://doi.org/10.1103/PhysRevB.47.558
- 17. Kresse, G. and Hafner, J. (1994) Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semi- conductor Transition in Germanium. Physical Review B, 49, 14251-14269. https://doi.org/10.1103/PhysRevB.49.14251
- 18. Kresse, G. and Furthmuller, J. (1996) Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Computational Materials Science, 6, 15-50. https://doi.org/10.1016/0927-0256(96)00008-0
- 19. Kresse, G. and Joubert, D. (1999) From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Physical Review B, 59, 1758-1775. https://doi.org/10.1103/PhysRevB.59.1758
- 20. Wang, Y., Gould, T., Dobson, J.F., Zhang, H.M., Yang, H.G., Yao, X.D. and Zhao, H.J. (2014) Density Functional Theory Analysis of Structural and Electronic Properties of Orthorhombic Perovskite CH3NH3PbI3. Physical Chemistry Chemical Physics, 16, 1424-1429. https://doi.org/10.1039/C3CP54479F
- 21. Dion, M., Rydberg, H., Schroder, E., Langreth, D.C. and Lundqvist, B.I. (2004) Van der Waals Density Functional for General Geometries. Physical Review Letters, 92, Article ID: 246401. https://doi.org/10.1103/physrevlett.92.246401
- 22. Klimes, J., Bowler, D.R. and Michaelides, A. (2011) Van der Waals Density Functionals Applied to Solids. Physical Review B, 83, Article ID: 195131. https://doi.org/10.1103/physrevb.83.195131
- 23. Baikie, T., Fang, Y.N., Kadro, J.M., Schreyer, M., Wei, F.X., Mhaisalkar, S.G., Graetzel, M. and White, T.J. (2013) Synthesis and Crystal Chemistry of the hybrid Perovskite (CH3NH3)PbI3 for Solid-State Sensitised Solar Cell Applications. Journal of Materials Chemistry A, 1, 5628-5641. https://doi.org/10.1039/c3ta10518k
- 24. Feng, J. and Xiao, B. (2014) Crystal Structures, Optical Properties, and Effective Mass Tensors of CH3NH3PbX3 (X = I and Br) Phases Predicted from HSE06. The Journal of Physical Chemistry Letters, 5, 1278-1282. https://doi.org/10.1021/jz500480m
- 25. Papavassiliou, G.C. and Koutselas, I.B. (1995) Structural, Optical and Related Properties of Some Natural Three- and Lower-Dimensional Semiconductor Systems. Synthetic Metals, 71, 1713-1714. https://doi.org/10.1016/0379-6779(94)03017-Z
- 26. Ishihara, T. (1994) Optical Properties of PbI-Based Perovskite Structures. Journal of Luminescence, 60-61, 269-274. https://doi.org/10.1016/0022-2313(94)90145-7
- 27. Mosconi, E., Amat, A., Nazeeruddin, M.K., Gratzel, M. and De Angelis, F. (2013) First-Principles Modeling of Mixed Halide Organometal Perovskites for Photovoltaic Applications. The Journal of Physical Chemistry C, 117, 13902- 13913. https://doi.org/10.1021/jp4048659
- 28. Lv, H.Z., Gao, H.W., Yang, Y. and Liu, L.K. (2011) Density Functional Theory (DFT) Investigation on the Structure and Electronic Properties of the Cubic Perovskite PbTiO3. Applied Catalysis A: General, 404, 54-58. https://doi.org/10.1016/j.apcata.2011.07.010
- 29. Pena, M.A. and Fierro, J.L.G. (2001) Chemical Structures and Performance of Perovskite Oxides. Chemical Reviews, 101, 1981-2018. https://doi.org/10.1021/cr980129f
- 30. Xing, G., Mathews, N., Sun, S., Lim, S.S., Lam, Y.M., Gratzel, M., Mhaisalkar, S. and Sum, T.C. (2013) Long-Range Balanced Electron- and Hole-Transport Lengths in Organic-Inorganic CH3NH3PbI3. Science, 342, 344-347. https://doi.org/10.1126/science.1243167