设为首页
加入收藏
期刊导航
网站地图
首页
期刊
数学与物理
地球与环境
信息通讯
经济与管理
生命科学
工程技术
医药卫生
人文社科
化学与材料
会议
合作
新闻
我们
招聘
千人智库
我要投搞
办刊
期刊菜单
●领域
●编委
●投稿须知
●最新文章
●检索
●投稿
文章导航
●Abstract
●Full-Text PDF
●Full-Text HTML
●Full-Text ePUB
●Linked References
●How to Cite this Article
Hans Journal of Computational Biology
计算生物学
, 2012, 2, 27-33
http://dx.doi.org/10.12677/hjcb.2012.23003
Published Online September 2012 (h
ttp://www.hanspub.org/journal/hjcb.html)
Molecular Dynamics Insight into the Interaction Mechanism
of Inhibitor PMI with MDMX
*
Weiyuan Cheng
1
, Zhiqiang Liang
2
, Wei Wang
2
, Changhong Yi
2
, Keyan Wang
2
, Hongyun Li
2
, Jianzhong Chen
2#
1
Affair Office, Shandong Jiaotong University, Jinan
2
School of Science, Shandong Jiaotong University, Jinan
Email:
#
chenjianzhong1970@163.com
Received: Aug. 23
rd
, 2012; revised: Aug. 27
th
, 2012; accepted: Sep. 3
rd
, 2012
Abstract:
Restoration of p53 function is considered to be a
new therapeutic strategy for anti-cancers. Molecular
Dynamics (MD) simulations
coupled with Molecular Me
chanics/Possion-Boltzman Surface Area (MM-PBSA) method
were used to study the mechanism of the PMI-MDMX interactio
n. The results show that van der walls energy drives the
PMI-MDMX interaction. Calculations based on residue-residue interaction were also performed, and the results not
only suggest that five residues of PMI can produce strong interaction with MDMX, but also the CH-CH, CH-
π
,
π
-
π
interactions predominate the binding of PMI in the hydr
ophobic cleft of MDMX. We expect that this study can
contribute significantly to the designs of the potent inhibitors inhibiting the PMI-MDMX interaction.
Keywords:
p53-MDMX Interaction; Molecular Dynamics; MM-PBSA; Binding Free Energy
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的
分子动力学研究
*
程伟渊
1
,梁志强
2
,王
伟
2
,伊长虹
2
,王克彦
2
,李洪云
2
,陈建中
2#
1
山东交通学院办公室,济南
2
山东交通学院理学院,济南
Email:
#
chenjianzhong1970@163.com
收稿日期:
2012
年
8
月
23
日;修回日期:
2012
年
8
月
27
日;录用日期:
2012
年
9
月
3
日
摘
要:
恢复抑癌蛋白
p53
的功能已经成为一种治疗癌症的新途径。本文采用分子动力学模拟和
MM-PBSA
方
法计算了抑制剂
PMI
与肿瘤蛋白
MDMX
的结合自由能。结果表明范德华相互作用驱动了
PMI
与
MDMX
的结
合。同时也使用基于残基对的自由能分解方法计算了残基–残基相互作用,结果不仅表明
PMI
的
5
个残基能与
MDMX
产生强烈的相互作用,而且也表明
CH-CH
,
CH-
π
,
π
-
π
相互作用主导了
PMI
在
MDMX
疏水性裂缝中的
结合。我们期望这个研究能为抑制
p53-MDMX
相互作用药物的研发提供理论上的启示。
关键词:
p53-MDMX
相互作用;分子动力学;
MM-PBSA
;结合自由能
1.
引言
人类
p53
蛋白是由
393
个氨基酸组成的核磷蛋
白,
p53
蛋白的一级结构可分为三个区域:
N
端酸性
区,由
1-75
位氨基酸组成;中间疏水区,由
100-300
位氨基酸组成;
C
端碱性区,主要由
310-390
位氨基
酸组成。在正常的细胞中,
p53
及其下游分子
MDM2
可以调控促癌转移分子
Slug
,通过形成
p53-MDM2-
Slug
复合体改变
Slug
的稳定性,抑制癌细胞转移能力
*
资助信息:国家自然科学基金
(Nos. 11104164)
、山东交通学院博士
启动资金和校自然基金。
#
通讯作者
。
Copyright © 2012 Hanspub
27
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
[1]
。因此蛋白
p53
素有“基因卫士”之称,它在细胞
生长周期和
DNA
修补等方面起到重要作用
[2]
。活性的
p53
能有效地抑制肿瘤的生长,保护人体肌体细胞免
受肿瘤的侵袭
[3]
。但是肿瘤蛋白
MDM2/MDMX
能直
接与
p53
结合,限制了
p53
的活性功能,世界上
50%
的癌症患者的体内发现了
MDM2/MDMX
的过度表达
证实这个结论
[4]
。因此,阻断
p53
与
MDM2 /M D MX
相互作用成为治疗癌症的新途径。
肿瘤蛋白
MDM2
和
MDMX
的
N
端与
p53
蛋白
N
端的
TA
区域结合,从而负调控
p53
的功能活性
[5]
。
MDMX
又称作
MDM4
,在氨基酸残基序列上,
MDMX
与
MDM2
的序列一致性达到
54%
[6,7]
,在结构上
MDMX
与
MDM2
显示了一个共同的
βαβαβ
的拓扑结
构
(
图
1)
[8]
。两个肿瘤蛋白都直接与
p53
相互作用,致
使
p53
功能失活。但是
MDMX
不能用作
p53
的转录
靶标,
这又在作用上不同于
MDM2
。研究表明
MDMX
在人体内的过度表达的确诱发了几种癌症
[9]
。因此抑
制
p53-MDM2
和
p53-MDMX
相互作用的双重抑制剂
的研发成为癌症治疗的重点。
抑癌蛋白
p53
疏水性表面的三个氨基酸残基
Phe19
′
,
Trp23
′
和
Leu26
′
插入到
MDM2
和
MDMX
的
疏水性裂缝
[5,10,11]
,从而与
MDM2/MDMX
相互作用。
在结构上,由
MDM2
的残基
His96
到
MDMX
的
Pro95
的转换致使
C
端
α
螺旋产生小的偏差
[8]
。因此一些课
题组以
p53-MDM2/MDMX
相互作用为靶标设计多个
类型的肽类和非肽类抑制剂
[3,12-14]
。这些抑制剂能以
较高的亲和能与
MDM2
结合,较好地抑制了
p53-
MDM2
相互作用,但对
p53-MDMX
相互作用抑制效
果比较差。
Pazgier
等人合成的肽类抑制剂
PMI
能与
MDMX
产生较高的结合能
[8]
。
PMI
在结构上呈现出
α
螺旋的形式
(
图
1)
,其残基序列为
TSFAEYWNLLSP
,
PMI
能够以
4.15 nM
的较强结合能力与
MDMX
结合
[8]
。因此在原子层次上研究
PMI
与
MDMX
的相互作
用机制对以
p53-MDMX
相互作用为靶标的抑制剂的
设计有重要意义。
目前,分子动力学模拟和结合自由能计算已经成
为研究蛋白质–蛋白质相互作用的重要工具。它们能
够在原子层次上理解
PMI
与
MDMX
的相互作用机
制,有利于阐明
PMI-MDMX
复合物的结构–亲和能
关系。本文选取了来自蛋白质库的结构
(3EQY)
[8]
作为
研究的初始模型。图
1
给出了
PMI-MDMX
复合物的
结构。本工作采用已经成功使用的
MM-PBSA
方法
[7,15-24]
和分子动力学模拟研究
PMI
与
MDMX
的结合
机制。能够为
p53-MDMX
相互作用的小分子抑制剂
的设计提供理论上的指导。
2.
材料和方法
2.1.
分子动力学模拟
用于
PMI-MDMX
复合物分子动力学模拟的初始
构象取自蛋白质库
(3EQY)
,所有的结晶水分子保留在
初始构象中。复合物晶体结构中缺失的氢原子由
Amber12
中的
Tleap
模块添加
[25]
。
MDMX
,
PMI
和水
分子的力场参数均取自
Amber
中的
ff03
力场
[26]
。
分子动力学模拟采用
Amber12
中的
Sander
程序。
PMI-MDMX
的复合体溶解在显性的
TIP3P
水盒子里,
水盒子的厚度为
10 Å
。两个氯离子添加到由水和复合
体组成的系统,以保证整个系统的电中性。为了消除
原子间一些不合理的接触,对复合体体系执行两步的
系统优化:
1)
约束溶质,优化溶剂和中和离子,约束
力常数为
100 kcal/(mol·Å
2
)
;
2)
无约束地优化整个系
统
.
每一步优化均先执行
3000
步的最都下降优化,接
着执行
3000
步的共轭梯度优化。然后在
300 ps
内把
系统从
0 K
加热到
300 K
,随后进行
300 ps
的常温
300
K
,常压
1
标准大气压的动力学平衡。最后是
10 ns
的无约束分子动力学模拟。
模拟期间采用
SHAK
方法限制所有含氢原子化
学键的伸缩
[27]
。模拟积分步长为
2 fs
。
PME
方法用来
计算长程静电相互作用。应用周期性边界条件以消除
Figure 1. Structure of PMI-MDMX complex. MDMX and PMI are
showed in new cartoon, the residues Phe3
′
, Trp7
′
and Leu10
′
are
displayed in ball-stick mode
图
1. PMI-MDMX
复合物的结构,
MDMX
和
PMI
以新卡通模式
显示,
Phe3
′
,
Trp7
′
和
Leu10
′
以球棒模式显示
Cop
yright © 2012 Hanspub
28
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
溶剂盒子的边缘效应。非成键相互作用的截断值为
2.2. MM-PBSA
计算
采用单轨迹方案的
MM-PBSA
方法计算肽类抑制
剂
PM
(1)
其中
10
.0 Å
。同时,监测了系统的能量和主链原子相对于
初始优化结构的
RMSD
随时间的函数,以判断系统平
衡的可靠性。
I
与
MDMX
的结合自由能。从动力学模拟轨迹
中每隔一定的时间间隔取出体系的结构,删掉构象中
的水分子和氯离子,对每一个构象由下面的方程计算
结合自由能。
1
MM so
GE GTS
M
M
E
是气相中的分子力学能贡献,
1
s
o
G
结合
溶解自
由能对
结合的贡献,
TS
表示熵变对 自由能
的贡献。
分
子
M
M
E
可以进一步分 部分:
成两
1
M
Mee
EEE
vdw
(2)
式中
和
分别表示气相中的静电相互作用和
1
ee
E
vdw
E
范德华能。溶解自由能也由如下的两个成分组成:
1
s
opbs
GGG
urf
3)
式中
(
p
b
G
和
s
urf
G
分别代表极性溶解自由能和非极
由能。前者的成性溶解自 分可以通过有限差分法求解
泊松–玻尔兹曼方程获得,溶质和溶剂的介电常数分
别设为
1.0
和
80.0
。后一项由下列的经验方程求解:
surf
GSASA
(4)
式中的值分别取为
0.00542 kcal/(mol·Å
2
)
和
0.92 kcal/
Δ
S
是由于自由度的变化导致的熵变对结合自由
能的贡献
(5)
其中
和
分别表示平动和转动自由度变化
简振模方
残基
–
残基相互作用的计算
自由能分解到每一个残基的贡献,有助于定量地
理解
G
mo
l.
T
,
TS
可以表述为方程
(5)
为:
trans rot vib
TS TSSS
trans
S
变化
b
rot
S
,这两项导致的熵 用经典的统计力学方法计算。
S
表示振动自由度的变化产生的熵贡献,该项应用
法进行计算。本文选择的体系中,抑制剂与
MDMX
的疏水性基团间产生较强 的吸 引作 用, 致使
结合前后运动的模式发生了变化。
vi
2.3.
抑制剂和残基的相互作用,有助于定量地阐明
PM
I-MDMX
复合体的结构亲和能关系。
Amber12
中
基于成对残基相互作用的自由能分解程序
GBSA
(Generalized Born/Surface Area)
能将残基–残基相互
作用分解为下面的方程表达式。
residue-residue 1
eevdw
GEE
pb surf
G
(6)
其中真空中的静电相互作用 和范德华
用
(
1
ee
E
)
作
(
vdw
E
)
由分子力学方法计算,极性溶剂化能
(
p
b
G
)
由
GB
型计算,而非极性溶剂化能
(
模
s
urf
G
)
由
LCPO
方法计算获得。
3.
结果和讨论
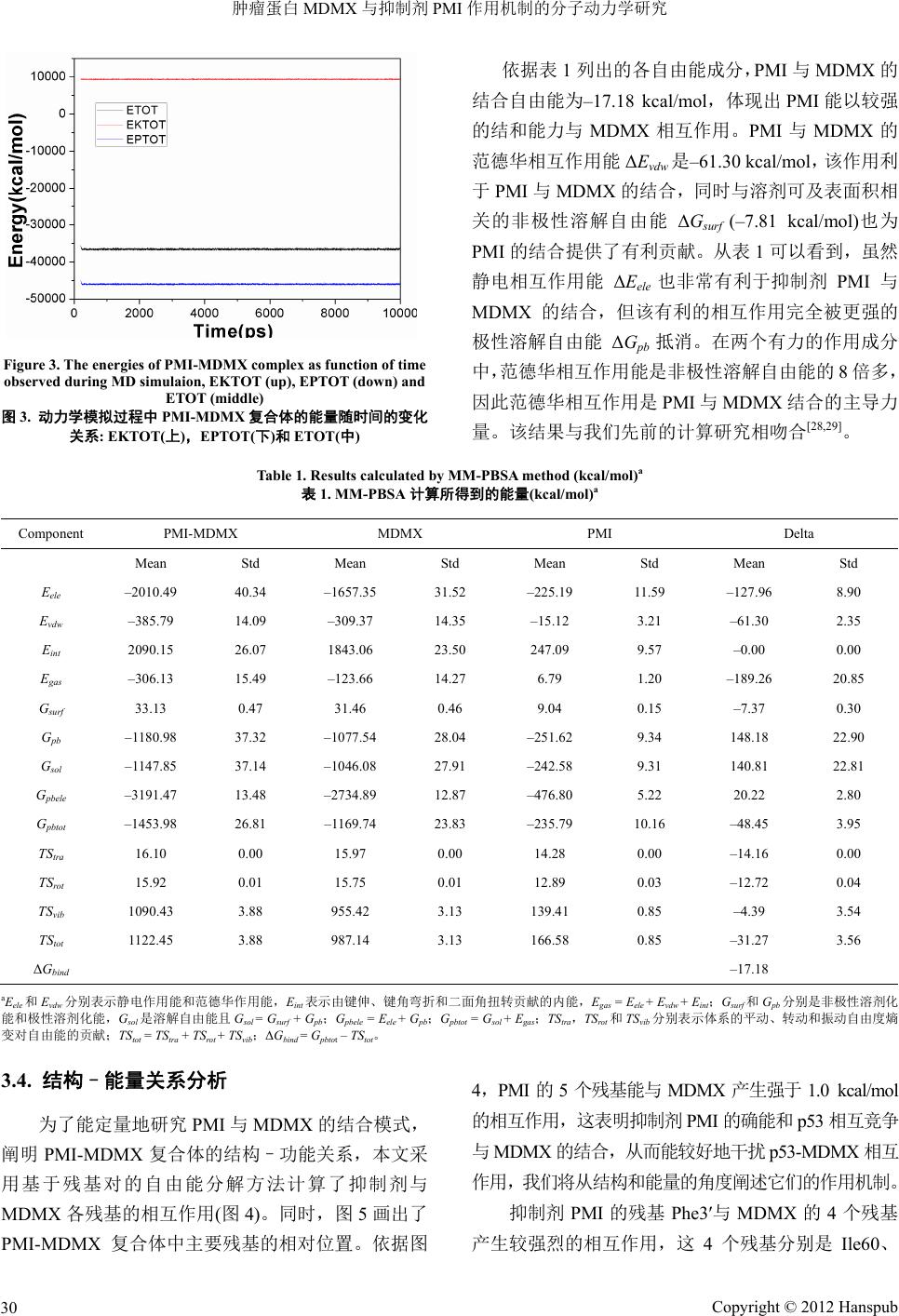
动力学平衡的稳定性
为评估动力学模拟的质量和系统平衡的可靠性
,
我们
3.2.
结合自由能计算
抑制剂与蛋白质的结合自由能不仅能衡量二者
结合
3.1.
使用
Am
ber
中的
Ptraj
模块分析了蛋白质主链原
子相对晶格结构的均方根偏差
(RMSD)
随时间的函数
(
图
2)
。依据图
2
,动力学模拟
4 ns
以后,系统达到了
平衡,且
RMSD
的平均值为
1.15 Å
,涨落范围低于
0.60 Å
。同时我们还监控了系统的动能、势能和总能
量随时间的变化关系
(
图
3)
,由图
3
观察到,模拟过程
中三种能量很稳定,无异常变化。以上结果表明用于
后加工分析的动力学轨迹的稳定性是可靠的。
的强
弱,也能反映出抑制剂与蛋白质结合的不同
作用成分。因此本文使用
MM-PBSA
方法计算了抑制
剂
PMI
和
MDMX
的结合自由能,自由能的组成成分
列在表
1
中。
Figure 2. Root-mean-square deviation of the backbone ats on om
PMI-MDMX complex
图
2. MD
模拟中主链原子的均方根偏差
Cop
yright © 2012 Hanspub
29
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
Copyright © 2012 Hanspub
30
Figure 3. The energies of PMI-MDMX complex as function of tim
observed during MD simulaion, EKTOT (up), EPTOT (down) an
关系
: EKTOT(
)
和
ETOT(
中
)
alculated by MM-PBSA method (kcal/mol)
a
表
1. MM-PBSA
计算所得到的能量
(kcal/mol)
a
Component PMI-MDMX I Delta
e
d
依据表
1
列出的各自由能成分,
PMI
与
MDMX
的
结合自由能为
–17.18 kcal/mol
,体现出
PMI
能以较强
的结和能力与
MDMX
相互作用。
PMI
与
MDMX
的
范德华相互作用能
Δ
E
vdw
是
–61.30 kcal/mol
,该作用利
于
PMI
与
MDMX
的结合,同时与溶剂可及表面积相
关的非极性溶解自由能
Δ
G
surf
(–7.81 kcal/mol)
也为
PMI
的结合提供了有利贡献。从表
1
可以看到,虽然
静电相互作用能
Δ
E
ele
也非常有利于抑制剂
PMI
与
MDMX
的结合,但该有利的相互 作用 完全 被更 强的
极性溶解自由能
Δ
G
pb
抵消。在两个有力的作用成分
中,范德华相互作用能是非极性溶解自由能的
8
倍多,
因此范德华相互作用是
PMI
与
MDMX
结合的主导力
量。该结果与我们先前的计算研究相吻合
[28,29]
。
ET
OT (middle)
图
3.
动力学模拟过程中
PMI-MDMX
复合体的能量随时间的变化
上
)
,
EPTOT(
下
Table 1. Results c
MDMX PM
Meatd Mean Std Mean Std Mean Std n S
E
– – 1 –
–
– – –
1
ele
2010.49 40.341657.35 31.52–225.191.59127.96 8.90
E
vdw
–385.79 14.09 –309.37 14.35 –15.12 3.21 –61.30 2.35
E
int
2090.15 26.07 1843.06 23.50 247.09 9.57 –0.00 0.00
E
gas
–306.13 15.49 –123.66 14.27 6.79 1.20 189.26 20.85
G
surf
33.13 0.47 31.46 0.46 9.04 0.15 –7.37 0.30
G
pb
1180.98 37.32 1077.54 28.04 251.629.34 148.18 22.90
G
sol
–1147.85 37.14 –1046.08 27.91 –242.58 9.31 140.81 22.81
G
pbele
–3191.47 13.48 –2734.89 12.87 –476.80 5.22 20.22 2.80
G
pbtot
–1453.98 26.81 –1169.74 23.83 –235.79 10.16 –48.45 3.95
TS
tra
16.10 0.00 15.97 0.00 14.28 0.00 –14.16 0.00
TS
rot
15.92 0.01 15.75 0.01 12.89 0.03 –12.72 0.04
TS
vib
090.433.88 955.42 3.13 139.41 0.85 –4.39 3.54
TS
tot
1122.45 3.88 987.14 3.13 166.58 0.85 –31.27 3.56
Δ
G
bind
–17.18
a
E
ele
和示静电作 范德华作用能,
E
int
表示由键伸、键角弯折和二面角扭转贡献的内能,
E
gas
=
E
ele
E
vdw
+
E
int
;
G
分别是非极 溶剂化
能和极性 能,
G
sol
是溶解自由能且
G
sol
=
G
surf
+
G
pb
;
G
pbele
=
E
ele
+
G
pb
;
G
pbtot
=
G
sol
+
E
gas
;
tra
,
TS
rot
和
TS
vib
分别表示体系的平 转动和振动自由度熵
MD
MX
的结合模式,
阐明
PMI-MDMX
复合体中主要残基的相对位置。依据图
4
,
PMI
的
5
个残基能与
MDMX
产生强于
1.0 kcal/mol
E
vdw
分别表
溶剂化
用能和
+
surf
和
G
pb
动、
性
TS
变对自由能的贡献;
TS
tot
=
TS
tra
+
TS
rot
+
TS
vib
;
Δ
G
bind
=
G
pbto
t
–
TS
tot
。
3.4.
结构
–
能量关系分析
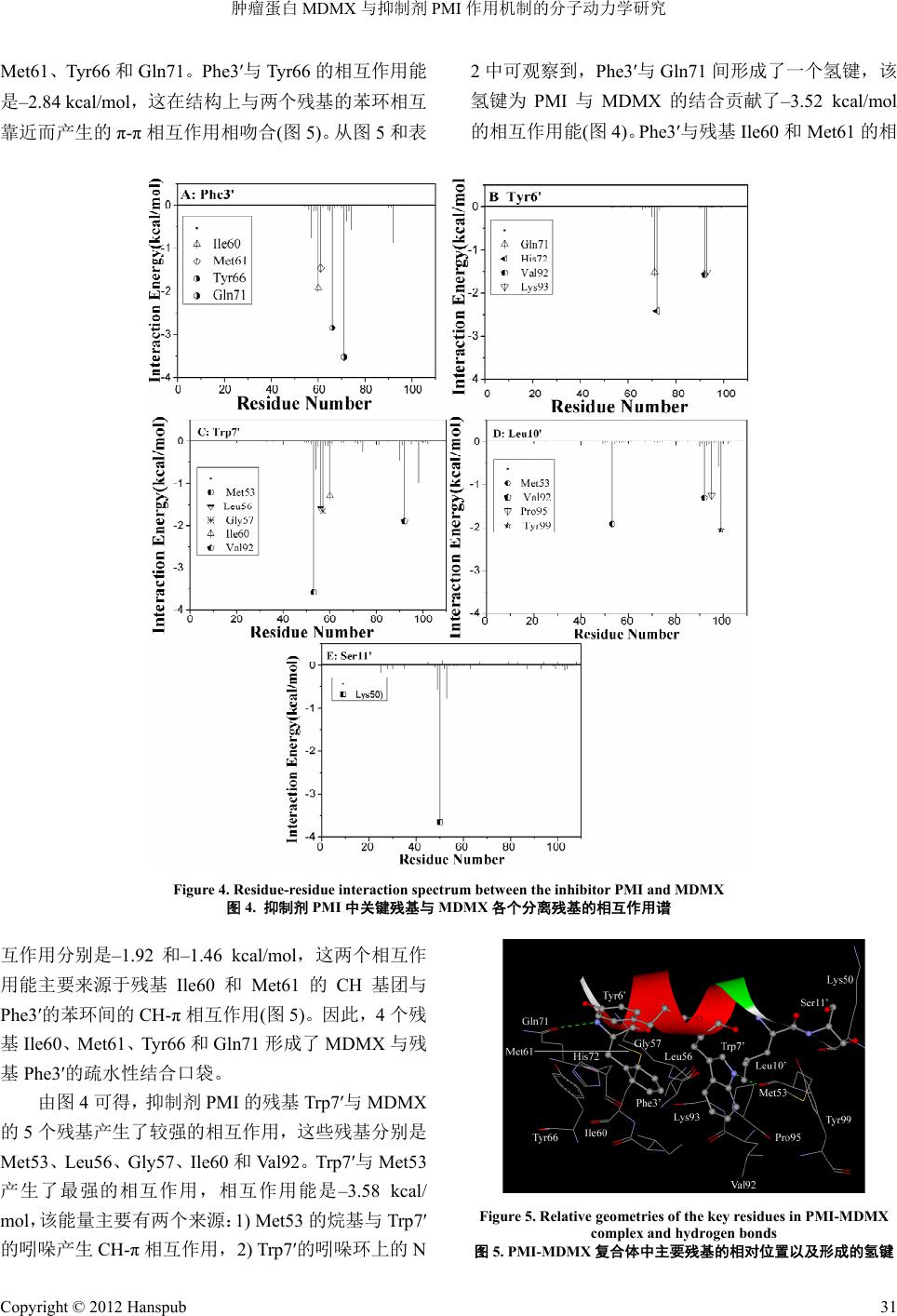
为了能定量地研究
PMI
与
PMI-MDMX
复合体的结构–功能关系,本文采
用基于残基对的自由能分解方法计算了抑制剂与
MDMX
各残基的相互作用
(
图
4)
。同时,图
5
画出了
的相互作用,这表明抑制剂
PMI
的确能和
p53
相互竞争
与
MDMX
的结合,从而能较好地干扰
p53-MDMX
相互
作用,我们将从结构和能量的角度阐述它们的作用机制。
抑制剂
PMI
的残基
Phe3
′
与
MDMX
的
4
个残基
产生较强烈的相互作用,这
4
个残基分别是
Ile60
、
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
Met61
、
Tyr66
和
Gln71
。
Phe3
′
与
Tyr66
的相互作用能
是
–2
.84 kcal/mol
,这在结构上与两个残基的苯环相互
靠近而产生的
π
-
π
相互作用相吻合
(
图
5)
。从图
5
和表
2
中可观察到,
Phe3
′
与
Gln71
间形成了一个氢键,该
氢键为
PMI
与
MDMX
的结合贡献了
–3.52 kcal/mol
的相互作用能
(
图
4)
。
Phe3
′
与残基
Ile60
和
Met61
的相
Figure 4. Residue-residue interaction spectrum between the inhibitor PMI and MDMX
图
4.
抑制剂
PMI
中关键残基与
MDMX
各个分离残基的相互作用谱
互作用分别是
–1.92
和
–1.46 kcal/mol
,这两个相互作
用能主要来源于残基
Phe3
′
的苯环间的
CH-
π
相互作用
(
图
5)
。因此,
4
个残
Val92
。
Tr p7
′
与
Met53
产生
Ile60
和
Met61
的
CH
基团与
基
Ile60
、
Met61
、
Tyr66
和
Gln71
形成了
MDMX
与残
基
Phe3
′
的疏水性结合口袋。
由图
4
可得,抑制剂
PMI
的残基
Trp7
′
与
MDMX
的
5
个残基产生了较强的相互作用,这些残基分别是
Met53
、
Leu56
、
Gly57
、
Ile60
和
了最强的相互作用,相互作用能是
–3.58 kcal/
mol
,该能量主要有两个来源:
1) Met53
的烷基与
Tr p7
′
的吲哚产生
CH-
π
相互作用,
2) Trp7
′
的吲哚环上的
N
Figure 5. Relative geometries of the key residues in PMI-MDMX
complex and hydrogen bonds
图
5. PMI-MDMX
复合体中主要残基的相对位置以及形成的氢键
Cop
yright © 2012 Hanspub
31
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
Table 2. The hydrogen bonds of the key residues
表
2.
主要残基的氢键分析
Donor
a
Acceptor
b
Distance
c
/Å Angle
d
/(
˚
)Freq.
e
/%
Phe3
′
-N-H Gln71-OE1 3.112 146.83 46.54
Tr p7
′
-NE1-HE1 Met53-OE1 2.754 151.43 92.25
a
供体原子;
b
受体原子;
c
距离;
d
角度;
e
占有率。
提供一个氢原子与
Met53
的羰基氧原子
(O)
形成一个
氢键
(
见表
2
和图
5)
。
Tr p7
′
与
Leu56
,
Gly57
,
Ile6
和
V
k
CH
基团 。故
MDMX
的残基
Met53
、
Leu56
、
Gly57
Ile60
和
Val92
形成
7
′
的疏
4
,基
′
与 的
残
V
5
r99
强
3
和
Val92
的相
互作用能分别是
–1.91
和
–11 kcal/mol
,从结构上看,
驱动了
PMI
在
MDMX
疏水性裂
缝中
53-MDM3/MDMX
相互作用双重抑制
供理论上的启示。
5.
致谢
W
1): 4-5.
[4]
S. Uldrijan, W. J. Pannekoek and K. H. Vousden. An essential
n of the extreme C-terminus of MDM2 can be provided
MX. The EMBO Journal, 2006, 26(1): 102-112.
[5]
G. M. Popowicz, A. Czarna and S. Wolf. Structures of low
iscovery.
[6]
T. L. Joseph, A. Madhumalar, C. J. Brown, et al. Differential
binding of p53 and nutlin to MDM2 and MDMX: Computational
167-1181.
t al. Computational studies of
difference ineptide and non-peptide inhibitors
M2 and
0
al92
相互作用能分别是
–1.58
,
–1.66
,
–1.29
和
–1.89
果恰好在结构上吻合了这
cal/mol
,该结
4
个残基的
与
Trp7
′
的吲哚环间的
CH-
π
相互作用
、
了
Trp
水性结合口袋。
依据图
PMI
的残
Leu10
MDMX
4
个
基
Met53
、
a l92
、
Pro9
和
Ty
形成 于
–1.0
kcal/mol
的相互作用。
Leu10
′
与
Met5
.3
这两个作用主要来自
Met53
和
Val92
的烷基与
Leu10
′
烷基间的
CH-CH
相互作用
(
图
5)
。
Leu10
′
与
Pr
o95
和
Tyr99
的相互作用能分别是是
–1.25
和
–2.06 kcal/mol
,
这在结构上与
Leu10
′
的烷基和
His96
的五元环和
Pro95
多元环间的
CH-
π
相互作用吻合
(
见图
5)
。因此
残基
Met53
、
Val92
、
Pro95
和
Tyr99
构建了
Leu10
′
的
疏水性结合口袋。
依据图
4
,除上述分析的三个残基外,还观察到
另外两个残基
Tyr6
′
和
Ser11
′
也与
MDMX
产生较强的
相互作用。
Tyr6
′
与残基
Gln71
、
His72
、
Val92
和
Lys93
的相互作用能分别是
–1.52
,
–2.42
、
–1.57
和
–1.52
kcal/mol
,这主要归功于
Tyr6
′
的苯环与残基
Gln71
、
His72
、
Val92
和
Lys93
烷基间的
CH-
π
相互作用。
Ser11
′
与
Lys50
的作用是
–3.68 kcal/mol
,这个作用主要来源
于
Se
r11
′
的烷基与
Lys50
烷基间的
CH-CH
相互作用。
上述分析基本上与
J. Z. H. Zhang
等人的量子力学研
究
[30]
以及我们先前的研究相吻合
[28,29]
。
总之,
CH-CH
,
CH-
π
和
π
-
π
相互作用
的结合。
4.
结论
本文对
PMI-MDMX
复合体执行了
10 ns
的分子
动力学模拟以研究抑制剂
PMI
与
MDMX
的作用机
制。采用
MM-PBSA
方法计算了
PMI
与
MDMX
的结
合自由能,结果表明范德华相互作用主导了抑制剂
PMI
与
MDMX
的结合。同时使用基于残基对的自由
能分解方法计算了残基–残基相互作用,结果显示
PMI
的
5
个残基
Phe3
′
、
Tyr6
′
、
Tr p7
′
、
Leu10
′
和
Ser11
′
能够与
MDMX
产生较强的相互作用。我们期望该研
究能够为抑制
p
剂的设计提
本文由国家自然科学基金
(No
s. 11104164)
、山东
交通学院博士启动资金和校自然基金资助。
参考文献
(References)
[1]
S. P. ang, W. L. Wang, Y. L. Chang, et al. p53 controls cancer
cell invasion by inducing the MDM2-mediated degradation of
Slug. Nature Cell Biology, 2009, 11(6): 694-704.
[2]
K. H. Vousden, D. P. Lane. p53 in health and disease. Nature
Reviews Molecular Cell Biology, 2007, 8(4): 275-283.
[3]
A. Shmueli, M. Oren. Regulation of p53 by Mdm2: Fate is in the
numbers. Molecular Cell, 2004, 13(
functio
by MD
molecular weight inhibitors bound to MDMX and MDM2 reveal
new approaches for p53-MDMX/MDM2 antagonist drug d
Cell Cycle, 2010, 9(6): 1104-1111.
studies. Cell Cycle, 2010, 9(6): 1
[7]
J. Chen, D. Zhang, Y. Zhang, e
binding modes of p
to MDM2/MDMX based on molecular dynamics simulations.
International Journal of Molecular Sciences, 2012, 13(2): 2176-
2195.
[8]
M. Pazgier, M. Liu, G. Zou, et al. Structural basis for high-
affinity peptide inhibition of p53 interactions with MD
MDMX. Proceedings of the National Academy of Sciences,
2009, 106(12): 4665-4670.
[9]
Y. F. M. Ramos, R. Stad, J. Attema, et al. Aberrant expression of
HDMX proteins in tumor cells correlates with wild-type p53.
Cancer Research, 2001, 61(5): 1839-1842.
[10]
J. Phan, Z. Li, A. Kasprzak, et
al. Structure-based design of high
affinity peptides inhibiting the interaction of p53 with MDM2
and MDMX. The Journal of Biological Chemistry, 2010, 285(3):
2174-2183.
[11]
A. Czarna, G. M. Popowicz, A. Pecak, et al. High affinity
interaction of the p53 peptide-analogue with human Mdm2 and
Mdmx. Cell Cycle, 2009, 8(8): 1176-1184.
[12]
K. Ding, Y. Lu, Z. Nikolovska-Coleska, et al. Structure-based
design of spiro-oxindoles as potent, specific
small-molecule
inhibitors of the MDM2-p53 interaction. Journal of Medicinal
Chemistry, 2006, 49(12): 3432-3435.
[13]
J. K. Murray, S. H. Gellman. Targeting protein-protein interactions:
Lessons from p53/MDM2. Biopolymers, 2007, 88(5): 657-686.
[14]
B. L. Grasberger, T. Lu, C. Schubert, et al. Discovery and
cocrystal structure of benzodiazepinedione HDM2 antagonists
that activate p53 in cells. Journal of Medicinal Chemistry, 2005,
Cop
yright © 2012 Hanspub
32
肿瘤蛋白
MDMX
与抑制剂
PMI
作用机制的分子动力学研究
Copyright © 2012 Hanspub
33
ynamic simulation.
namics study. Chemistry—A European Journal, 2008,
ease complexed with
t into mechanism of small
predicting the binding mode to HIV-1 R
utational
odel1. Journal of Molecular Biology, 2000,
ergy calculations.
tions of symmetric fluoro-
hibitors binding to Hsp90 using molecular
A computational analysis of
ulation. Compu-
48(4): 909-912.
[15]
J. Chen, S. Zhang, X. Liu, et al. Insights into drug resistance of
mutations D30N and I50V to HIV-1 protease inhibitor TMC-114:
Free energy calculation and molecular d
Journal of Molecular Modeling, 2010, 16(3): 459-468.
[16]
E. L. Wu, K. L. Han and J. Z. H. Zhang. Selectivity of neutral/
weakly basic P1 group inhibitors of thrombin and trypsin by a
molecular dy
14(28): 8704-8714.
[17]
T. Hou, R. Yu. Molecular dynamics and free energy studies on
the wild-type and double mutant HIV-1 prot
amprenavir and two amprenavir-related inhibitors: Mechanism
for binding and drug resistance. Journal of Medicinal Chemistry,
2007, 50(6): 1177-1188.
[18]
J. Chen, J. Wang, B. Xu, et al.
Insigh
molecule inhibitors of the MDM2-p53 Interaction: Molecular
dynamics simulation and free energy analysis. Journal of Molecular
Graphics and Modelling, 2011: 46-53.
[19]
J. Wang, P. Morin, W. Wang, et al. Use of MM-PBSA in
reproducing the binding free energies to HIV-1 RT of TIBO
derivatives and T of
interaction mechanisms of peptide and non-peptide inhibitors
with MDMX based on molecular dynamics sim
efavirenz by docking and MM-PBSA. Journal of the American
Chemical Society, 2001, 123(22): 5221-5230.
[20]
J. Wang, R. M. Wolf, J. W. Caldwell, et al. Development and
testing of a general amber force field. Journal of Comp
Chemistry, 2004, 25(9): 1157-1174.
[21]
W. Wang, P. A. Kollman. Free energy calculations on dimer
stability of the HIV protease using molecular dynamics and a
continuum solvent m
303(4): 567-582.
[22]
J. Chen, M. Yang, G. Hu, et al.
Insights into the functional role
of protonation states in the HIV-1 protease-BEA369 complex:
Molecular dynamics simulations and free en
Journal of Molecular Modeling, 2009, 15(10): 1245-1252.
[23]
J. Z. Chen, M. Y. Yang, C. H. Yi, et al. Molecular dynamics
simulation and free energy calcula
substituted diol-based HIV-1 protease inhibitors. Journal of
Molecular Structure: THEOCHEM, 2009, 899(1-3): 1-8.
[24]
C. H. Yi, J. Z. Chen, S. H. Shi, et al. A computational analysis of
pyrazole-based in
dynamics simulation and the
MM-GBSA method. Molecular
Simulation, 2010, 36(6): 454-460.
[25]
D. A. Case, T. A. Darden, T. E. Cheatham III, et al. AMBER 12.
University of California, San Francisco, 2012.
[26]
Y. Duan, C. Wu, S. Chowdhury, et al. A point-charge force field
for molecular mechanics simulations of proteins based on
condensed-phase quantum mechanical calculations. Journal of
Computational Chemistry, 2003, 24(16): 1999-2012.
[27]
T. G. Coleman, H. C. Mesick and R. L. Darby. Numerical
integration. Annals of Biomedical Engineering, 1977, 5(4): 322-
328.
[28]
W. Cheng, J. Chen, Z. Liang, et al.
tational and Theoretical Chemistry, 2012, 984: 43-50.
[29]
程伟渊
,
梁志强
,
张庆刚等
. p53-MDM2
相互作用的分子力学
和动力学研究
[J]
.
原子与分子物理学报
, 2012, 29(3): 393-
399.
[30]
Y. Ding, Y. Mei and J. Z. H. Zhang. Quantum mechanical studies
of residue-specific hydrophobic interactions in p53-MDM2 binding.
The Journal of Physical Chemistry B, 2008, 112(36): 11396-
11401.