Modern Physics
Vol.05 No.03(2015), Article ID:15233,7
pages
10.12677/MP.2015.53008
Effect of Nanochannel Properties on the Pressure of Fluid Film by Molecular Dynamics
Yan Jia, Ye Xue
Xi’an Polytechnic University, Xi’an Shaanxi
Email: jiayan05@126.com
Received: Apr. 28th, 2015; accepted: May 12th, 2015; published: May 18th, 2015
Copyright © 2015 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Molecular dynamics method is applied to study the influence of potential interaction strength between the liquid and the solid on the properties of fluid film in nanochannels. The results indicate: the pressure distribution of fluid film is changed in Nanochannels with the change of the height of channel in the outlet end and the liquid-solid potential interaction strength. At the same time, the jet phenomenon can occur in the outlet end. The difference of the pressure distribution between the results obtained by molecular dynamics simulation and that by NS or Renolds equation is much bigger. At this point, the jet phenomenon is more obvious. The jet velocity increases linearly as the liquid-solid potential rises. With the increasing height of the outlet, the nanoscale effect becomes weaker and weaker. The pressure profile gradually approaches to that of the macro-flow. The liquid-solid potential has no significant effect on the jet velocity.
Keywords:Molecular Dynamics, Potential Interaction Strength, Fluid Film, Jet
流道特性对流体压力影响的分子动力学研究
贾 妍,薛晔
西安工程大学,陕西 西安
Email: jiayan05@126.com
收稿日期:2015年4月28日;录用日期:2015年5月12日;发布日期:2015年5月18日

摘 要
应用分子动力学方法研究纳米流道不同固–液间相互作用势能指数对液体流动特性的影响。研究结果显示,纳米尺度下流道出口高度及固液间相互作用势能不同会引起流体压力的改变,并出现射流现象。出口端距离较小时,流道内液体流动规律不再服从NS方程——按照分子动力学模拟与按NS或Renolds方程求解得到的压力分布结果存在较大的差异,此时射流现象比较明显,射流速度随着固液间势能指数的增强呈线性增大趋势。随着出口端高度的增加,纳米尺度效应减弱,压力分布逐渐趋近宏观流动下的压力分布曲线,固液间势能指数对于射流速度的大小不再产生明显的影响。
关键词 :分子动力学,势能指数,液体薄膜,射流

1. 引言
宏观状态下当运动表面正常运转时,两个固体壁面间液体的流动特性可以通过NS方程或Reynolds方程研究。但随着微纳技术在众多科学领域的广泛应用,纳米尺度下流体的流动特性是否依然符合经典的流体动力学理论已经受到人们越来越广泛的重视。当物体尺寸达到纳米级以后其性质会发生重大的改变,此时用传统的连续介质理论研究纳米尺寸下流体的流动特性可能已不再有效,而从统计物理学中衍生出来的分子动力学模拟,由于不需要引入经典流体动力学的连续介质假设,以微观粒子间的相互作用势为基础可以研究尺寸极小的系统,因此在探索纳米尺寸下物质特性方面有着独到的优势。
Shin-Pon Ju [1] 研究了Au纳米粒子周围水的粘附性,结果显示在Au纳米粒子很小的状态下对水的有很强的作用力。Xie Hui和Liu Chao [2] 的研究结果显示固体与液体间表现为亲水性时,固体表面附近温度对流道内的液体分子数有着重要的影响。Hai Hoang和Guillaume Galliero [3] 利用分子动力学研究了固体壁面特性对液体结构的影响,研究结果显示在壁面的有些地方会产生液体与固体之间的粘合。Kiran Sasikumar和Pawel Keblinsk [4] 使用分子动力学研究在热的固体粒子束附近的空穴现象。Itsuo Hanasaki等人 [5] - [7] 利用液体活塞(FPM)模型研究了液体在碳纳米管内的流动。结果显示,在碳纳米管内部流体的速度分布出现了差异,靠近壁面的液体的流速小于内部的流速。并从分子角度探索了宏观领域中存在的射流现象的微观机理。
然而,目前无论对于宏观领域内的研究还是微观领域内液体薄膜的研究都是针对两固体壁面平行条件下液体薄膜行程以后,而对于存在于一般机械系统中的两接触壁面分离过程中流体薄膜在纳米尺度下流动演化特性的研究很少涉及。本文采用分子动力学的方法主要研究两个固体壁面分离过程中形成的非平行纳米流道内不同固液间相互作用势指数对液体薄膜特性的影响。
2. 模拟系统与基本原理
2.1. 模拟系统
模拟系统由固体壁面和壁面间的液体分子构成,如图1所示。为了研究方便模拟过程中保持上壁面静止不动,让下壁面沿x方向产生相对运动。
模拟过程中边界条件和分子插入原则按照ITSUO [5] - [7] 应用分子动力学方法研究液体活塞作用下碳
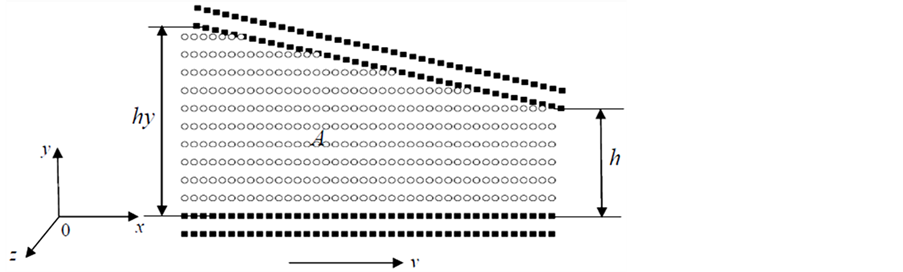
Figure 1. Sketch of the simulation system. ○: Liquid Molecular, ■: Solid Molecular
图1. 模拟系统示意图。○表示流体分子,■表示固体壁面分子
纳米管内液体的流动特性和水的射流现象时选取的边界条件和分子插入原则。仅z方向采用周期性边界条件。插入分子的速度按照初始温度根据麦克斯韦–玻尔兹曼分布随机确定;插入分子的位置在采取随机确定的同时还要遵循以下的原则:
首先,当初始条件下模拟区域内的分子数确定时,靠近入口端处单层分子晶格中所包含的分子的数目即为初始条件下尝试插入的分子数目。并且尝试插入的数目随着模拟区域中分子数目和壁面运动速度的变化而变化。当模拟区域的分子数增加时,认为尝试插入的分子数减小;当模拟区域内的分子数小于初始条件对应的分子数时,尝试插入的分子数增加。每时间步尝试插入的分子数遵循以下的方式:
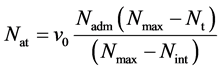 (1)
(1)
其中:Nat表示每时间步尝试插入的分子数;v0表示下壁面运动速度;Nadm表示初始状态外部区间每时间步长尝试插入分子数;Nmax表示模拟区间最大容纳分子数;Nt表示模拟区间瞬时分子数;Nint表示初始状态模拟区间内能容纳分子数。
模拟区域最大容纳的分子数Nmax根据YAO [8] 提出的方法确定。当一定体积内的分子按照密排面心立方晶格排布时,此时固定体积内的分子数最大。
其次,分子的尝试插入过程中还应满足MIHALY [9] 提出的分子插入原则。当液体分子尝试进入模拟区域时,只有当模拟区域存在一个液体分子所占的空间时,液体分子才能插入模拟区域成为模拟区域内的分子。
为了研究模拟区域内液体沿x方向不同层面特性的变化,将模拟区域沿x方向划分成若干个小的子区域。由于模拟中两固体壁面处于非平行状态,为了方便各小区域内压强的比较,小区域的划分遵循等体积原则。
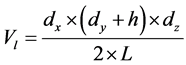 (2)
(2)
其中:Vl表示小层面的体积;dx,dy,dz表示模拟区域沿x,y和z方向尺寸;h表示两固体壁面间出口端的距离;L表示划分小区域的个数。
各个小层面压力计算公式 [10] - [12] :
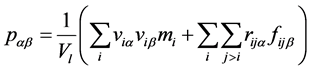 (3)
(3)
压力计算公式中,第一项表示分子动能对压力作用效果,第二项表示粒子间相互作用势能对压力的作用效果。
其中:α,β表示分别表示x,y和z方向;viα表示第i个粒子沿α方向的速度分量;viβ表示第i个粒子沿β方向的速度分量;mi表示第i个粒子的质量;rijα表示第i与第j个粒子间距离沿α方向分量;fijβ表示第i与第j个粒子间相互作用力沿β方向分量。
2.2. 势能作用模型
模拟系统中的液体采用液态氩,液态氩分子间的相互作用势采用Lennard-Jones (LJ) [10]
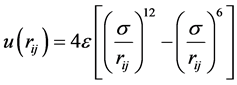 (4)
(4)
其中:rij表示第i个分子与第j个分子之间的距离;σ表示分子特征长度,也是分子的直径;ε表示分子间相互作用强度的能量特征值。
整个模拟过程采用无量纲参数,具体约化单位处理见表1。
为反映液体与固体之间不同作用势能对液体润滑膜的影响效果,分子动力学模拟选取的液体和固体壁面之间的LJ相互作用势为 [13]
 (5)
(5)
其中:usl表示液体分子和固体分子间的LJ势能;εsl表示液体分子和固体分子之间的作用势能;σsl表示液体分子和固体分子之间的距离参数;c表示用来调整液体分子和固体壁面之间的相互作用效果,即浸润性,其意义是指液体分子与固体壁面分子之间的作用力相对于距离相等的液体分子和固体分子作用力的倍数,c越大则表示液体分子和壁面固体分子之间的作用势能越强,c = 1则表示液体分子和固体分子之间的势能作用。本文中为了方便计算选取固液间分子的作用势能和距离参数等于液体分子的作用势能和距离参数。

Table 1. Units of the various quantities
表1. 分子动力学模拟中物理量的约化单位
2.3. 计算方法
对液体进行分子动力学模拟研究时液体分子的初始位置不会改变模拟系统最终的特性,原则上对于液体任意的初始分布都是合理的 [14] ,这里初始时刻流体分子按照简单的面心立方晶格(FCC)结构排列;分子间的距离按照给定的密度值确定。分子的初始速v0按照初始温度根据麦克斯韦–玻尔兹曼分布随机给出。
模拟过程中纳米流道入口端的宽度保持不变均为hy = 9.35,系统在x和z方向的计算域尺寸分别为34.8和5.1。h为纳米流道出口端不同分离阶段两个固体壁面间的距离。
当粒子初始位置,速度和粒子上的作用力确定后,根据牛顿运动定律,液体分子的运动方程为: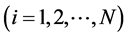
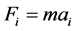 。通过积分运动方程,可以得出任意时刻液体分子的速度和位置,积分过程选用蛙跳法 [10] [14] [15] 。积分过程中的时间步长Dh = 0.005τ,为节约计算时间,在计算分子间作用力时,取截断半径rc = 2.5σ,即当两个粒子间距离大于截断半径时,其相互作用可以忽略不计。
。通过积分运动方程,可以得出任意时刻液体分子的速度和位置,积分过程选用蛙跳法 [10] [14] [15] 。积分过程中的时间步长Dh = 0.005τ,为节约计算时间,在计算分子间作用力时,取截断半径rc = 2.5σ,即当两个粒子间距离大于截断半径时,其相互作用可以忽略不计。
为研究液体与固体壁面间不同势能作用指数对模拟区域内液体流动特性的影响。模拟中保持下壁面以稳定的速度v0 = 1.0运行,液体与固体壁面间势能作用的强弱通过势能作用指数c进行调节。本文采用的几种典型固液间势能作用指数分别为0.2、0.5、0.7、0.9、1.0、2.0、3.0和4.0。
3. 模拟结果
3.1. 流道内流体压力分布曲线
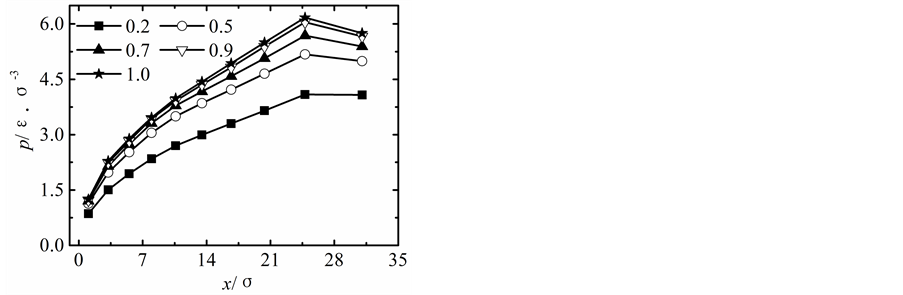
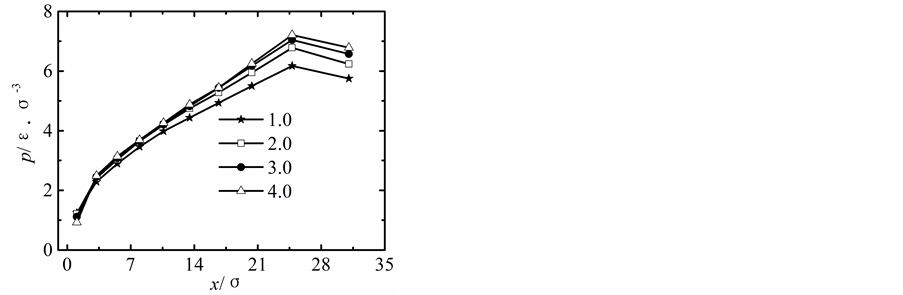
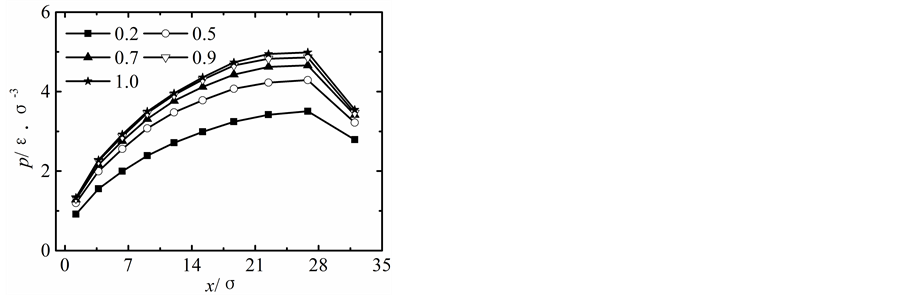
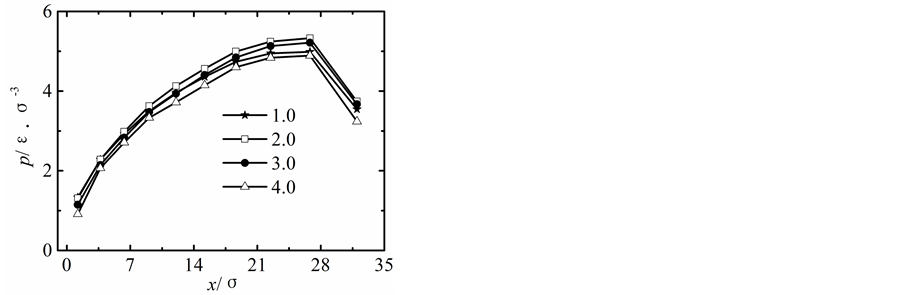
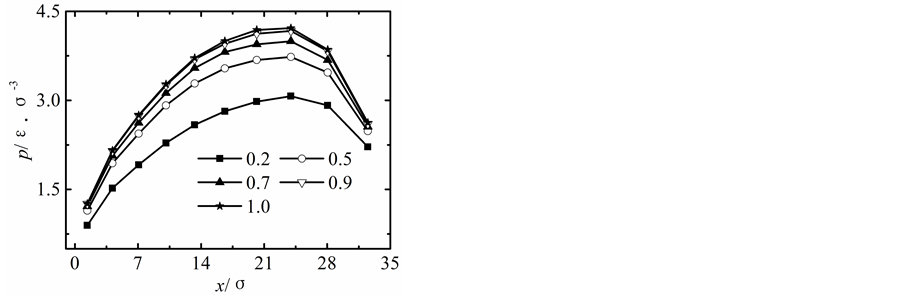
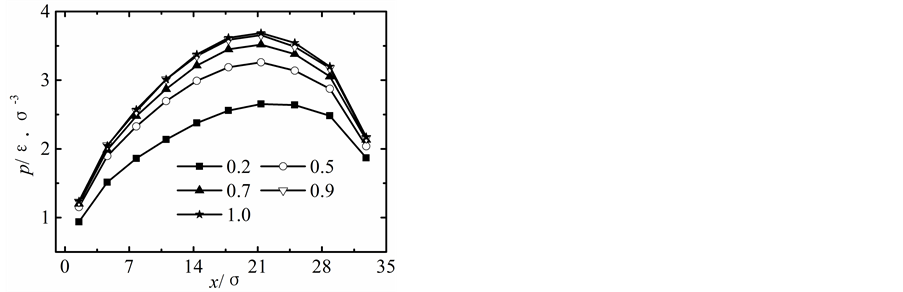
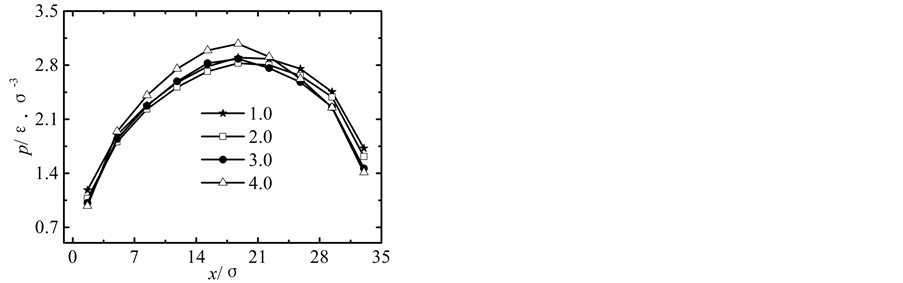
在保持纳米流道入口端距离不变的情况下改变出口端距离,获得出口端不同状态下所对应的沿x方向液体压力随固液间相互作用势能指数的变化曲线,如图2所示。
当出口端的距离较小时,模拟获得的液体的压力分布曲线与宏观状态下通过NS方程求得的液体压力分布曲线存在较大差异,随着纳米流道出口端距离的增加,模拟区域内液体压力的最大值不断下降,并随固体壁面间的距离的增加越来越偏离出口端,同时液体压力的变化曲线越来越平缓,当h > 5.1时,模拟区域内液体压力分布曲线与宏观状态下的压力分布曲线基本相符合。
纳米流道内,固液间相互作用势能指数较弱时,其对液体压力的影响较大,当c < 1.0时,随着固液间相互作用势能指数的增加,模拟区域内液体的压力不断升高;当c > 1.0时,固液间势能作用指数较强时模拟区域内液体压力分布基本不受固液间相互作用势能指数增加的影响。引起这种现象的主要原因是由于当c < 1.0时,固液间相互作用势能指数较弱,液体在固体表面表现为疏水性,固体壁面运动运动过程中壁面的固体分子始终对液体分子的运动产生影响;当c > 1.0时,固液间相互作用势能指数较强,液体在固体表面表现为亲水性,固体壁面运动过程中由于固体分子与液体分子的强烈作用会使固体表面附着较多的液体分子,在分子相互间力的作用范围内实际表现为液体分子之间的相互作用,此时固液间相互作用势能指数的增加对液体压力的影响基本保持不变。
纳米流道出口端不同状态下,模拟区域内液体最大压力随固液间势能指数变化曲线,如图3所示。
当h = 1.7时,随着固液间势能指数的增加,液体压力的最大值不断增加;当h > 3.4,c < 1.0时,对于固液间相互作用势能指数较弱的情况,随着固液间势能作用指数的增强不断增加;但当c > 1.0时,固液间相互作用势能指数改变对液体最大压力的影响基本可以忽然不计。
3.2. 射流速度
纳米流道出口端距离改变时,射流速度随固液间相互作用势能指数的变化曲线,如图4所示。
当h = 1.7时,由于出口端两固体壁面间的距离较小,固液间势能作用强弱对射流速度的影响较为明显,随着势能指数从0.2增加到4.0,出口端的射流速度呈线性的增加。
 (a)
(a)
 (b)
(b)
 (c)
(c)
 (d)
(d)
 (e)
(e)
Figure 2. Pressure profiles. (a) h = 1.7 liquid-solid potential interaction strength = 0.2 - 4.0, (b) h = 3.4 liquid-solid potential interaction strength c = 0.2 - 4.0, (c) h = 5.1 liquid-solid potential interaction strength c = 0.2 - 4.0, (d) h = 6.8 liquid-solid potential interaction strength c = 0.2 - 4.0, (e) h = 8.5 liquid-solid potential interaction strength c = 0.2 - 4.0
图2. 压力分布曲线。(a) h = 1.7固液间势能指数c = 0.2~4.0,(b) h = 3.4固液间势能指数c = 0.2~4.0,(c) h = 5.1固液间势能指数c = 0.2~4.0,(d) h = 6.8固液间势能指数c = 0.2~4.0,(e) h = 8.5固液间势能指数c = 0.2~4.0
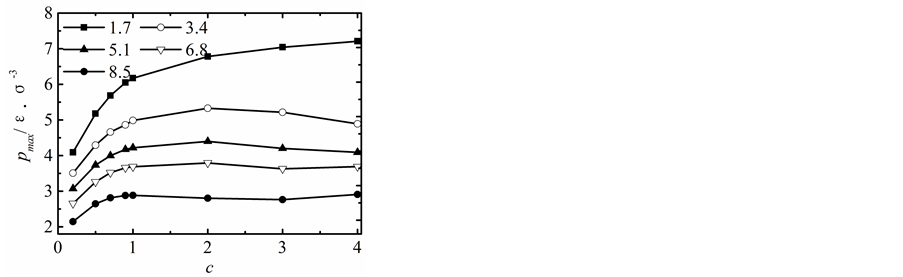
Figure 3. Maximum pressure profiles
图3. 最大压力变化曲线
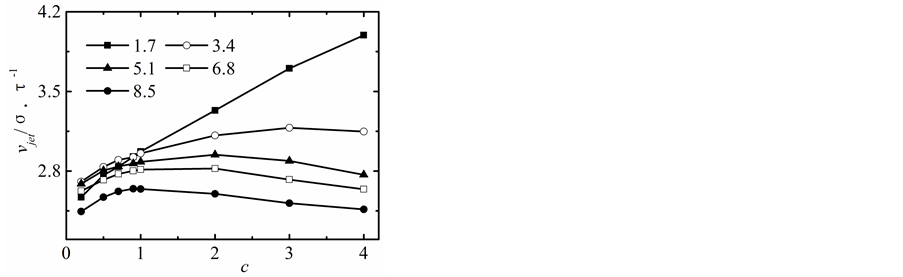
Figure 4. Jet velocities
图4. 射流速度
随着纳米流道出口端距离的增加,当h = 3.4时,固液间较强作用势能指数对射流速度的影响迅速下降,对于c > 1.0,固液间势能作用指数越强,射流速度下降的幅度越大;对于c < 1.0,射流速度随固液间势能作用指数的增加基本没有发生改变。当纳米流道出口端的距离h > 5.1时,射流速度随固液间势能指数的增加不再发生明显的改变。
综上所述,当纳米流道出口端的距离较小,固液间势能作用指数对射流速度的影响较大,随着固液间势能指数的增加,射流速度基本呈线性增加。随着纳米流道出口端分离距离的增加射流速度不断减小,尤其对于固液间较强的势能作用指数,射流速度下降的幅度较大。当进出口的差距较小的时,固液间势能作用指数的强弱对射流速度影响看越来越小。值得注意的是,较强的固液相互作用势能下,液体的射流速度反而出现微弱的下降趋势。
4. 结论
1) 当纳米流道出口端距离较小时,靠近出口端的压力较大,纳米流道内液体压力分布不均匀;随着出口端高度的增加,纳米尺度效应逐渐减弱,压力分布越来越趋近宏观流动下的压力分布曲线。
2) 针对纳米流道内液体的流动状态由出口端封闭向楔形小间隙流动演化过程中,存在射流现象的分子动力学研究表明,纳米流道出口端的距离较小时,随着固液间势能指数的增加,射流速度基本呈线性增加;出口端的距离较大时,射流速度随固液间势能指数的增加不再发生明显的改变。
基金项目
西安工程大学博士启动基金(No. BS1204)。
文章引用
贾 妍,薛 晔, (2015) 流道特性对流体压力影响的分子动力学研究
Effect of Nanochannel Properties on the Pressure of Fluid Film by Molecular Dynamics. 现代物理,03,57-64. doi: 10.12677/MP.2015.53008
参考文献 (References)
- 1. Ju, S.-P. (2005) A molecular dynamics simulation of the adsorption of water molecules surrounding an Au nanoparticle. The Journal of chemical Physics, 122, 094718(1-6).
- 2. Xie, H. and Liu, C. (2012) Molecular dynamics simulation of gas flow in nanochannel with a Janus interface. AIP Advances, 2, 042126(1-8).
- 3. Hoang, H. and Galliero, G. (2013) Shear behavior of a confined thin film influence of the molecular dynamics scheme employed. The Journal of Chemical Physics, 138, 054707(1-11).
- 4. Sasikumar, K. and Keblinski, P. (2014) Molecular dynamics investigation of nanoscale cavitation dynamics. The Journal of Chemical Physics, 141, 234508(1-7).
- 5. Hanasaki, I. and Nakatani, A. (2006) Flow structure of water in carbon nanotubes: Poiseuille type or plug-like? The Journal of Chemical Physics, 124, 144708(1-9).
- 6. Hanasaki, I. and Nakatani, A. (2006) Fluidized piston model for molecular dynamics simulations of hydrodynamic flow. Modelling and Simulation in Materials Science and Engineering, 14, s9-s20.
- 7. Hanasaki, I. and Nakatani, A. (2009) Molecular dynamics of a water jet from carbon nanotube. Physical Review, E79, 046307(1-7).
- 8. Yao, J. (1982) Monte carlo simulation of the grand canonical ensemble. Molecular Physics, 46, 587-594.
- 9. Mihaly, M. (1980) A cavity-biased (T,V, μ) Monte Carlo method for the computer simulation of fluids. Molecular Physics, 40, 901-906.
- 10. Allen, M.P. and Tildesley, D.J. (1987) Computer simulation of liquids. Oxford University Press, New York.
- 11. Camara, L.G. and Bresme, F. (2003) Molecular dynamics simulations of crystallization under confinement at triple point conditions. Journal of Chemical Physics, 19, 2792-2800.
- 12. Fernamdo, B and Nicholas, Q. (1999) Computer simulation of wetting and drying of spherical perticulates at a liquid- vapor interface. Journal of Chemical Physics, 10, 3536-3547.
- 13. 曹炳阳 (2005) 速度滑移及其对微纳尺度流动影响的分子动力学研究. 博士论文, 清华大学, 北京.
- 14. Frenkel, D. and Smit, B. (2002) Understanding molecular simulation from algorithms to applications. Academic Press, California.
- 15. Rapaport, D.C. (2004) The art of molecular dynamics simulation. Cambridge University Press, New York.