Applied Physics
Vol.06 No.04(2016), Article ID:17407,5
pages
10.12677/APP.2016.64009
Study of Crystal Structure, Electronic Structure and Magnetic Properties of CaMnO3 from Hybrid Functional Approach
Jiahong Dai*, Tianyi Cai, Sheng Ju
College of Physics, Optoelectronics and Energy, Soochow University, Suzhou Jiangsu

Received: Apr. 4th, 2016; accepted: Apr. 22nd, 2016; published: Apr. 25th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Based on the Heyd-Scuseria-Ernzerhof (HSE) screened hybrid density functional theory, we have studied the crystal structure, electron structure, and magnetic properties of perovskite transition metal oxide CaMnO3. Compared with traditional local density approximation and generalized gradient approximation, the crystal structure within HSE is in good agreement with experimental data. In addition, a 2.5 eV band gap is close to 3.1 eV from experiment. Furthermore, with the magnetic exchange coupling constants, the magnetic transition Néel temperature of CaMnO3is 87 K, consistent with the experiment value of 131 K. These findings indicate that the hybrid functional could provide a proper description of insulating transition metal oxides.
Keywords:First Principles, CaMnO3, Screened Hybrid Density Functional

CaMnO3晶体结构、电子结构和磁性质的 杂化泛函研究
戴佳洪*,蔡田怡,雎胜
苏州大学,物理与光电∙能源学部,江苏 苏州

收稿日期:2016年4月4日;录用日期:2016年4月22日;发布日期:2016年4月25日

摘 要
基于杂化密度泛函理论,我们研究了钙钛矿型过渡金属氧化物CaMnO3的晶格结构和电子结构,并与传统的局域密度近似和广义梯度近似相比较。计算表明杂化泛函给出的晶体结构与实验结果非常吻合,同时2.5 eV大小的能隙与实验上测得的3.1 eV也十分接近。更进一步,我们计算了Mn4+离子间的磁交换作用系数,并用蒙特卡罗方法估算出体系的尼尔温度为87 K,这与实验上的131 K也比较符合。这些结果表明杂化泛函可以很好预测绝缘型过渡金属氧化物的物理性质。
关键词 :第一性原理,CaMnO3,杂化密度泛函理论

1. 引言
近年来,过渡金属氧化物由于其丰富的物理性质受到人们的广泛关注。尤其是在锰氧化中发现的庞磁电阻效应,更是引发了科学家对钙钛矿型过渡金属氧化物的研究兴趣 [1] [2] 。同时锰氧化物由于其电荷,自旋,晶格和轨道等多自由度之间的耦合,表现出丰富的物理特性,如金属绝缘转变、轨道序、多铁性和磁电效应等。在其构造的异质界面上,研究者发现了更为奇异的母体材料所不具备的特性 [3] - [5] 。
第一性原理计算方法在预测材料性质方面起着非常重要的作用。虽然传统的基于局域密度近似(LDA)和广义梯度近似(GGA)的计算方法可以较好的预测材料基态的物理性质,但计算得到的带隙与实验并不符合。最近人们已经应用杂化泛函理论来研究一些氧化物的物理性质,包括VO2、V2O3、Ti2O3、LaTiO3和YTiO3等,并且且得到了与实验比较吻合的结果 [6] 。对于锰氧化物而言,基于杂化泛函的第一性原理研究还比较少。本文以CaMnO3为例,利用杂化泛函理论研究了其块材的晶体结构、电子结构和磁性质,并且与传统的LDA和GGA计算方法相比较,我们发现杂化泛函的计算方法不仅可以准确描述CaMnO3块材的晶体结构和磁结构,而且得到了与实验更为接近的带隙。这为我们进一步研究过渡金属氧化物的物理性质提供了依据。
2. 计算方法
我们的计算运用基于投影缀加平面波的方法(PAW)的第一性原理软件包VASP [7] [8] 。我们分别用LDA、GGA和基于广义梯度近似下的杂化泛函方法Heyd-Scuseria-Ernzerhof (HSE) [9] 计算了CaMnO3块材的晶格结构,电子结构和磁性质。我们的计算主要考虑Ca的3s32p64s2价电子(10个),Mn的3p63d54s2 (13个),O的2s2p4 (6个)的贡献,平面波截断能都是500 eV,LDA和GGA的Monkhorst-Packk点是6 × 4 × 6,HSE的k点是4 × 3 × 4。晶体结构和电子结构优化收敛标准为10−5 eV。本文在屏蔽库仑场的HSE计算中,用Hartree-Fock(HF)交换函数替换部分短程的(sr)PBE交换函数,其交换关联函数表示如下:
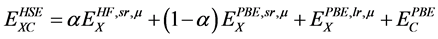 (1)
(1)
其中μ是库仑屏蔽系数,α是HF掺杂的比例。我们的杂化泛函的计算方法是HSE06方法,即α和μ分别为0.25和0.2。
3. 计算结果与讨论
CaMnO3和LaMnO3类似,具有Pnma的钙钛矿结构,如图1所示。由于Mn-O和Ca-Mn之间的杂化作用,氧八面体略有钆铁氧型的倾斜,和杨–泰勒畸变。平面内的Mn-O键分成长键l和短键s,垂直方向的Mn-O键成为m。因为两种过渡金属Mn原子的电子构型不同,CaMnO3的Q2模和Q3模比LaMnO3小的多。CaMnO3中的Mn4+离子电子结构是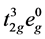 ,同一自旋方向上的t2g轨道正好被满占,抑制了杨-泰勒效应。使G类反铁磁绝缘态成为其基态。
,同一自旋方向上的t2g轨道正好被满占,抑制了杨-泰勒效应。使G类反铁磁绝缘态成为其基态。
我们运用HSE,GGA和LDA的计算方法,分别计算了G类反铁磁态(平面内和平面间都是反铁磁排列),A类反铁磁态(平面内是铁磁排列,平面间是反铁磁排列),C类反铁磁态(平面内是反铁磁排列,平面间是铁磁排列),普通铁磁态体系的总能量,见表1。三种计算方法的结果都表明,CaMnO3体系的磁基态为G类反铁磁态。
虽然HSE,PBE和LDA的计算结果都显示,G类反铁磁态是CaMnO3的磁基态,但是三者在晶体结构的计算上有着显著的区别。我们将不同方法计算得到的晶体结构参数与Bozin等人的实验数据比较,见表2,可以明显的发现HSE的计算结果与实验更为吻合。
对材料晶格常数和体积的计算结果比较可以发现,GGA的计算结果偏大,而LDA的计算结果偏小,只有HSE的计算结果与实验测量值十分吻合。HSE计算的优势同样还体现在氧八面体的键长和键角等方面。因此与GGA和LDA相比,在材料晶格结构的预测方面,HSE有着巨大的优势。
此外,我们还探究了CaMnO3的电子结构,并且分析了不同计算结果。Jung等人通过光谱实验,测量出CaMnO3的带隙为3.1 eV [11] 。CaMnO3中的Mn离子为正四价,有3个电子占据在3 d轨道上。体系在eg轨道和t2g轨道间打开能隙。我们分别计算了HSE,PBE和LDA的总态密度,见图2,带隙分别是2.51 eV,0.69 eV和0.57 eV。我们可以明显的看到,HSE在体系带隙的计算上优势非常明显。与GGA和LDA相比,HSE得到的带隙与实验结果十分吻合。
我们运用海森堡模型计算了体系中磁性原子平面内(ac)最近邻的交换作用J1,平面间(沿b轴)最近邻J2和次近邻J3的交换作用,见表3。海森堡模型的哈密顿量表示如下:
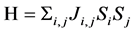 (2)
(2)
其中Ji,j是磁交换常数,Si和Sj是电子自旋。此外,我们还运用蒙特卡洛方法模拟出体系的磁转变温度TN。

Figure 1. Crystal structure of the CaMnO3. Left is side view and right is top view
图1. CaMnO3的晶体结构示意图,左边为侧视图,右边为俯视图

Table 1. Total energy of CaMnO3 for different magnetic orderings
表1. CaMnO3各种磁结构的总能量
Table 2. Comparison between experimental (10 K) and calculated structural parameters of CaMnO3
表2. 实验(10 K)测得CaMnO3的晶体结构参数和计算所得到的CaMnO3晶体结构参数
Table 3. The magnetic exchange coupling parameters and Néel temperature of CaMnO3
表3. CaMnO3磁交换作用系数和尼尔温度

Figure 2. Total density of states of CaMnO3
图2. CaMnO3的总态密度
我们发现只有HSE和PBE的计算结果和实验上的测量值131 K [12] 比较接近,LDA的预测值(290 K)与实验值相差甚远。
4. 总结
基于杂化密度泛函的第一性原理计算,我们研究了钙钛矿型过渡金属氧化物CaMnO3的晶格结构,电子结构和磁性质。我们发现HSE可以很好的描述CaMnO3的物理特性。相比LDA近似和GGA近似,HSE计算得到的带隙与实验值更为接近。进一步利用海森堡模型并结合蒙特卡罗方法,我们发现体系的反铁磁转变尼尔温度为87 K,这也与实验上所测得的131 K比较吻合。通过这些的研究,我们认为基于HSE的第一性原理计算方法在描述过渡金属氧化物的物理性质方面有着巨大优势。
基金项目
国家自然科学基金(批准号:11104193,11374220)资助的课题。
文章引用
戴佳洪,蔡田怡,雎 胜. CaMnO3晶体结构、电子结构和磁性质的杂化泛函研究
Study of Crystal Structure, Electronic Structure and Magnetic Properties of CaMnO3 from Hybrid Functional Approach[J]. 应用物理, 2016, 06(04): 63-67. http://dx.doi.org/10.12677/APP.2016.64009
参考文献 (References)
- 1. Von Helmolt, R., Wecker, J., Holzapfel, B., Schultz, L. and Samwer, K. (1993) Giant Negative Magnetoresistance in Perovskitelike La2/3Ba1/3MnOx Ferromagnetic Films. Physical Review Letter, 71, 2331. http://dx.doi.org/10.1103/PhysRevLett.71.2331
- 2. Salamon, M.B. and Jaime, M. (2001) The Physics of Manganites: Structure and Transport. Review of Modern Physics, 73, 583. http://dx.doi.org/10.1103/RevModPhys.73.583
- 3. Dong, S., Yu, R., Yunoki, S., Alvarez, G., Liu, J.M. and Dagotto, E. (2008) Magnetism, Conductivity, and Orbital Order in (LaMnO3)2n/(SrMnO3)n Superlattices. Physical Review B, 78, 201102. http://dx.doi.org/10.1103/PhysRevB.78.201102
- 4. Hou, F., Cai, T.Y., Ju, S. and Shen, M.R. (2011) Magnetic Reconstruction at Oxygen-Deficient SrMnO3(001) Surface: A First-Principle Investigation. Applied Physics Letter, 99, 192510. http://dx.doi.org/10.1063/1.3659492
- 5. Hou, F., Cai, T.Y., Ju, S. and Shen, M.R. (2012) Half-Metallic Ferromagnetism via the Interface Electronic Reconstruction in LaAlO3/SrMnO3 Nanosheet Superlattices. ACS Nano, 6, 8552. http://dx.doi.org/10.1021/nn303943t
- 6. Iori, F., Gatti, M. and Rubio, A. (2012) Role of Nonlocal Exchange in the Electronic Structure of Correclated Oxides. Physical Review B, 85, 115129. http://dx.doi.org/10.1103/PhysRevB.85.115129
- 7. Blochl, P.E. (1994) Projector Augmented-Wave Method. Physical Review B, 50, 17953. http://dx.doi.org/10.1103/PhysRevB.50.17953
- 8. Kresse, G. and Furthmüller, J. (1996) Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Computational Materials Science, 6, 15. http://dx.doi.org/10.1016/0927-0256(96)00008-0
- 9. Heyd, J., Scuseria, G.E. and Ernzerhof, M. (2003) Hybrid Functionals Based on a Screened Coulomb Potential. Journal of Chemical Physics, 118, 827.
- 10. Bozin, E.S., Sartbaeva, A., Zheng, H., Wells, S.A., Mitchell, J.F., Proffen, Th., Thorpe, M.F. and Billinge, S.J.L. (2008) Structure of CaMnO3 in the Range 10 K ≤ T ≤ 500 K from Neutron Time-of-Flight Total Scattering. Journal of Physics and Chemistry of Solids, 69, 2146-2150. http://dx.doi.org/10.1016/j.jpcs.2008.03.029
- 11. Jung, J.H., Kim, K.H., Eom, D.J., Noh, T.W., Choi, E.J., Yu, J., Kwon, Y.S. and Chung, Y. (1997) Determination of Electronic Band Structures of CaMnO3 and LaMnO3 Using Optical-Conductivity Analyses. Physical Review B, 55, 15489. http://dx.doi.org/10.1103/PhysRevB.55.15489
- 12. Neumeier, J.J. and Cohn, J.L. (2000) Possible Signatures of Magnetic Phase Segregation in Electron-Doped Antiferromagnetic CaMnO3. Physical Review B, 61, 14319. http://dx.doi.org/10.1103/PhysRevB.61.14319