Nuclear Science and Technology
Vol.
10
No.
01
(
2022
), Article ID:
47773
,
8
pages
10.12677/NST.2022.101003
Zr-H相互作用计算模拟研究与应用
陈嘉威,张智鑫,马雁*
华北电力大学,北京
收稿日期:2021年11月8日;录用日期:2021年11月26日;发布日期:2022年1月7日

摘要
压水堆中锆包壳的氢化破损会严重影响反应堆的安全运行,吸氢、氢扩散、氢化物沉淀等过程都有可能引发锆包壳的氢脆断裂。本文主要研究了运用密度泛函理论、第一性原理、分子动力学及相场法模拟Zr-H相互作用的方法,并通过Zr-H2O体系建模及分子动力学方法模拟了界面处的Zr-H相互作用过程。
关键词
压水堆,锆包壳,Zr-H相互作用,Zr-H2O体系,分子动力学模拟

Research and Application of Zr-H Interaction Simulation
Jiawei Chen, Zhixin Zhang, Yan Ma*
North China Electric Power University, Beijing
Received: Nov. 8th, 2021; accepted: Nov. 26th, 2021; published: Jan. 7th, 2022

ABSTRACT
The hydrogen damage of the zirconium cladding in PWR will seriously affect the safe operation of the reactor. The processes of hydrogen absorption, hydrogen diffusion and hydride precipitation may cause hydrogen embrittlement fracture of the zirconium cladding. This paper mainly studies the methods of using the density functional theory, first principles, molecular dynamics and phase field methods to simulate the Zr-H interaction, and through Zr-H2O system modeling and molecular dynamics simulation, the Zr-H interaction process at the interface was simulated.
Keywords:PWR, Zirconium Cladding, Zr-H Interaction, Zr-H2O System, Molecular Dynamics Simulation

Copyright © 2022 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
作为燃料包壳材料,Zr合金要在严苛工况中服役 [1] [2],一回路中的Zr-H2O反应将导致H的溶解和渗透 [3] [4] [5],而Zr合金中过量的H会形成脆性氢化物 [6] [7] 并影响包壳管的机械性能。计算模拟方法能在微观尺度对Zr中H的原子间相互作用和微观结构演变进行模拟,因此本文研究了运用密度泛函理论、第一性原理、分子动力学及相场法模拟Zr-H间相互作用的方法,并应用分子动力学(MD)方法自主建模并模拟了Zr-H2O体系反应过程,模拟界面处的Zr-O化学键形成过程及H扩散过程,为探究Zr-H2O界面处的Zr-H相互作用机理提供新的研究思路。
2. Zr-H相互作用计算模拟
2.1. H的吸收和扩散
已有的计算模拟结果显示,H原子优先占据四面体(TE)位 [8],且H与空位之间存在很强的相互吸引作用 [9]。hcp结构的Zr间隙位置如图1所示。
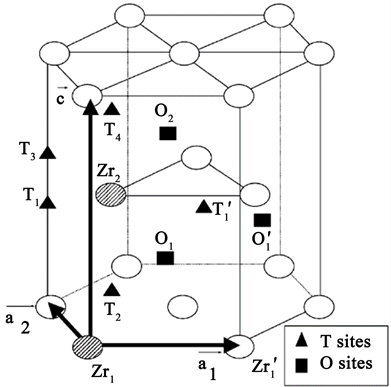
Figure 1. Labelling of interstitial sites in hcp Zr [8]
图1. hcp结构的Zr中的间隙位置 [8]
C. Domain等人 [8] 使用密度泛函理论研究了Zr-H系统的性质,通过模拟得知H原子在低温下优先占据四面体(T)位置,且H在扩散时优先沿八面体(O)位置进行,并且在基面中交替进入T位置和O位置。P.A.T. Olsson等人 [9] 通过密度泛函理论研究了氢的吸收对Zr力学性能的影响,发现氢填充的空位有助于降低表面能以及增加不稳定的堆垛层错能,使锆合金的延展性降低,且由于存在氢填充空位,断裂功和峰值应力降低。
Christopher I等人 [10] 运用LAMMPS代码进行分子动力学计算,Zr-H作用势由Christensen等人 [11] 基于EAM电位功能形式在这项工作中开发的原子间电位表示的,势函数具有以下形式:
(1)
其中,D, 和 是参数, 的值设定为5.0。通过计算得出了H在Zr中的扩散率,并与实验的测定值高度吻合,并且发现H与Zr中的空位相互作用会促进空位聚集,而H也会大大降低Zr中空位的迁移率。
2.2. ZrxHy的形成
Xueyan Zhu等人 [12] 运用第一性原理系统研究了ZrHx (x = 0.5, 1, 1.5, 2)的结构以及它们的热力学性质,发现了 结构的ZrH0.5的新稳定结构,讨论了氢化锆在不同温度下的稳定性,并提出了 的相变反应路径。计算过程中,Zr1-xHx的形成焓算式为:
(2)
其中, 、 和 分别为各物质对应的总能量。
Yongfeng Zhang等人 [13] 使用LAMMPS软件模拟了 氢化物的形成机理。由于描述Zr-H系统原子间相互作用的势函数中COMB势是最合适的 [14],因此Yongfeng Zhang等人使用COMB势 [15] 描述原子间相互作用。使用分子动力学计算了H/Zr比在1.0到2.0之间变化的各种氢化物的形成能(如图2),若所有四面体点位都被占据,则会生成 氢化物,通过随机除去H原子,可以分别以1.6和1.0的H/Zr比获得 和 氢化物,由于具有COMB电位,氢化物的fcc结构都被认为是最稳定结构,因此生成的氢化物可被视为具有各种H/Zr比的 氢化物。
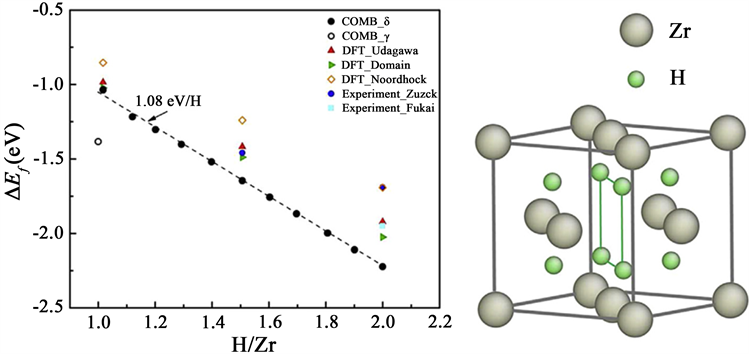
Figure 2. Hydride formation energy per ZrHX molecule as a function of the H/Zr ratio (left) and the crystal structure of hydrides used in the calculations (right) [13]
图2. ZrHx分子形成能随H/Zr比的变化趋势(左)和计算所用氢化物的结构(右) [13]
Ludovic Thuinet等人 [16] 通过密度泛函理论原理的VASP软件对Zr合金中的氢化物进行了原子级的研究,发现 -氢化物的形成可以从 -hcp或 氢化物开始,具体形成途径取决于H含量。而Yongfeng Zhang等人通过计算模拟进一步提出了 氢化物的两种可能形成的途径:1) ;2) 。两条路径都涉及hcp-fcc的相变,且最终产物都是 氢化物。
原子尺度的计算可以很好地模拟氢扩散和氢化物析出,而中尺度氢化物组织则可以通过微结构尺度的相场模拟来实现 [17]。相场模型已用于研究几种氢化物相的微观结构演变:Thuinet研究了 氢化物的微观结构的演变 [18];L. Thuinet等人 [19] 和X.Q. Ma等人 [20] 模拟了晶粒结构;外加应力情况下的 氢化物形态也由相场模拟实现 [21] [22] [23],例如X.Q. Ma等人 [21] 利用相场动力学模型研究了 -氢化物在锆中的沉淀和生长,通过数值求解结构变量的时间相关Ginzburg-Landau方程和浓度变量的Cahn-Hilliard扩散方程,从而确定空间相关场变量的时间演化,研究应力对氢化锆体系成核,生长和粗化过程的影响;Tae WookHeo等人 [24] 在近期的研究中开发了一个综合相场模型,他们所开发的模型可用于模拟包含多个晶粒 -Zr中氢化物的形成,并研究了 -Zr中的 氢化物在金属晶体中的体积膨胀现象。由下图3对比可以明显看到,相场模拟可很大程度上仿真模拟出真实情况。

Figure 3. Comparison of (a) TEM observation by Bailey [25] (b) Phase field simulation from Shi and Xiao [22]
图3. 比较(a) Bailey [25] 的TEM观察结果与(b) Shi和Xiao [22] 的模拟结果
2.3. Zr-H相互作用计算模拟方法对比
密度泛函理论仅能模拟几百个原子和短时间尺度,分子动力学模拟能模拟更大体系,并能温度和压强的共同影响;第一性原理能够通过计算实现对ZrHx的结构及其稳定性、热力学性质、氢化物相变反应路径的研究,而分子动力学能给出不同温度下的扩散系数,得到化学键断裂、结合等化学过程;相场法适用于中尺度微观模拟,可对微米量级的氢化物沉淀、生长与体膨胀现象进行计算,可与相同微观尺度的实验结果进行对比分析,模拟尺度及计算重点与分子动力学不同。应用分子动力学方法可在微观尺度下更好地模拟出一定压力、温度下Zr-H2O界面处的Zr-H相互作用,并能模拟Zr-O键的形成及H扩散过程。
3. MD模拟Zr-H2O界面处的Zr-H相互作用
Zr-H-O原子间的相互作用力决定Zr-H2O体系的微观结构与物理、化学性质,因此选用合适的原子间相互作用势是该分子动力学模拟的关键。最初认为模拟Zr-H原子间相互作用最合适的势函数是Christensen等人 [11] 基于EAM电位功能改进的EAM电势,然而EAM势并不能形成原子间化学键,因此该势函数不具备模拟Zr-H体系化学反应的能力。经过进一步调研 [13] [14] [15] 与计算模拟,确定COMB势可以完全描述Zr-H-O系统,且支持各原子间化学键的形成与断裂,适用于Zr-H2O体系的分子动力学模拟。其势函数形式为:
(3)
因此Zr-H2O体系的分子动力学模拟选用COMB3势展开。
在Zr-H2O体系模型box中,hcp结构的Zr位于box下方,H2O分子位于box上方,box尺寸为44.79 Å × 45.26 Å × 74.18 Å,H、O、Zr原子总数量设置为3884个,盒子底部是c = 5.147,a = 3.233,c/a = 1.592的hcp结构Zr基体,上部真空中随机分布100个水分子如图4所示。

Figure 4. Zr-H2O calculation model box
图4. Zr-H2O计算模型box
运用旋转矩阵R使模拟box中的H2O分子满足位置随机分布。x、y、z方向上的旋转矩阵与欧拉角旋转矩阵R如下式(4)~(7)所示:
(4)
(5)
(6)
(7)
设定box为周期性边界条件,运用NPT系综,在300~600 K的温度梯度下,运用共厄梯度法cg进行能量最小化的弛豫过程中,热力学参数时间步长2e-3ps,归一化热动力学输出值。使得到温度、电荷、压力、体积、原子位置均方位移及动势能,从微观角度研究不同工况下对锆水反应中H原子扩散的影响。
Zr-H2O体系在15 MPa、600 K的初始环境下,水分子全部分布在Zr基体(绿色)上方(图5(a)),随着模拟的进行,水分子中的H-O键(H原子为蓝色,O原子为红色)发生断裂(图5(b)),O吸附到Zr表面会形成Zr-O键,H继续向基体内扩散(图5(c))。采用共邻分析(CNA)方法,结果显示使c/a = 1.592的hcp结构数量从3136下降到2439。
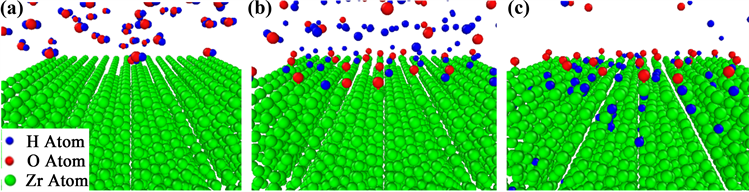
Figure 5. Zr-H2O system (a) No reaction, (b) H-O bond fracture, (c) H atom diffusion into Zr matrix
图5. Zr-H2O体系(a) 未反应,(b) H-O键断裂,(c) H原子扩散进Zr基体间隙
在运行压力15 MPa、运行温度300~600 K的工况下,随着Zr-H2O反应的持续进行,H的扩散系数D及扩散现象如图6所示,结果显示随着温度升高H扩散系数D逐渐提高。结合图6(a)~(d),可观察到温度的提高会加速H原子扩散到Zr基体,说明温度提高使锆水反应向正反应方向移动,使Zr基体体系在NPT系综下体积膨胀。

Figure 6. The H atomic diffusion coefficient of NPT at 15 MPa and different temperatures (a) 300 K, (b) 400 K, (c) 500 K, (d) 600 K
图6. NPT系综15 MPa下各温度的H原子扩散系数(a) 300 K,(b) 400 K,(c) 500 K,(d) 600 K
4. 结论
本文研究了微观尺度下Zr-H体系的计算模拟方法,得出以下结论:
1) 密度泛函理论在研究Zr中H的间隙位填充方式时具有明显优势,具备计算H在Zr中间隙固溶规律的能力。
2) 第一性原理能够通过计算实现对ZrHx的结构及其稳定性、热力学性质、氢化物相变反应路径的研究。
3) 分子动力学能够在原子尺度模拟H在Zr中的聚集与迁移行为,并能模拟Zr-H-O体系间的化学反应。
4) 相场法适用于中尺度微观模拟,可对微米量级的氢化物沉淀、生长与体积膨胀现象进行计算,可与相同微观尺度的实验结果进行对比分析。
5) 建立Zr-H2O体系反应模型,使用分子动力学方法成功模拟了界面处的锆水反应,形成Zr-O键,并模拟了H在Zr中的扩散行为,为原子尺度Zr-H2O界面处的Zr-H相互作用模拟提供了一种可行的技术手段。
基金项目
大型压水堆破损燃料检测及性状分析技术研究(KZ19011083)。
文章引用
陈嘉威,张智鑫,马 雁. Zr-H相互作用计算模拟研究与应用
Research and Application of Zr-H Interaction Simulation[J]. 核科学与技术, 2022, 10(01): 20-27. https://doi.org/10.12677/NST.2022.101003
参考文献
- 1. 史丽生. 锆及锆合金的吸氢[C]//中国核学会. 中国核科学技术进展报告(第二卷)——中国核学会2011年学术年会论文集第4册(核材料分卷、同位素分离分卷、核化学与放射化学分卷). 北京: 中国核学会, 2011: 8.
- 2. 杨文斗. 反应堆材料学[M]. 北京: 原子能出版社, 2000.
- 3. Suman, S., Kaleem Khan, M., Pathak, M., Singh, R.N. and Chakravartty, J.K. (2015) Hydrogen in Zircaloy: Mechanism and Its Impacts. International Journal of Hydrogen Energy, 40, 5976-5994. https://doi.org/10.1016/j.ijhydene.2015.03.049
- 4. Lewis, M.B. (1984) Deuterium-Defect Trapping in Ion-Irradiated Zirconium. Journal of Nuclear Materials, 125, 152- 159. https://doi.org/10.1016/0022-3115(84)90542-7
- 5. Vizcaı́no, P., Banchik, A.D. and Abriata, J.P. (2002) Solubil-ity of Hydrogen in Zircaloy-4: Irradiation Induced Increase and Thermal Recovery. Journal of Nuclear Materials, 304, 96-106. https://doi.org/10.1016/S0022-3115(02)00883-8
- 6. Wang, Z.Q., Li, Y.H., Li, Z.Z., Zhou, H.B. and Lu, G.H. (2019) Investigating Behavior of Hydrogen in Zirconium by First-Principles: From Dissolution, Diffusion to the Interaction with Vacancy. Nuclear Instruments and Methods in Physics Research Section B, 458, 1-6. https://doi.org/10.1016/j.nimb.2019.07.036
- 7. Motta, A.T., Capolungo, L., Chen, L.Q., Cinbiz, M.N., Daymond, M.R., Koss, D.A., Lacroix, E., Pastore, G., Simon, P.C.A., Tonks, M.R., Wirth, B.D. and Zikry, M.A. (2019) Hydrogen in Zirconium Alloys: A Review. Journal of Nuclear Materials, 518, 440-460. https://doi.org/10.1016/j.jnucmat.2019.02.042
- 8. Domain, C., Besson, R. and Legris, A. (2002) Atomic-Scale Ab-Initio Study of the Zr-H System: I. Bulk Properties. Acta Materialia, 50, 3513-3526. https://doi.org/10.1016/S1359-6454(02)00173-8
- 9. Olsson, P.A.T., Kese, K. and Alvarez Holston, A.M. (2015) On the Role of Hydrogen Filled Vacancies on the Embrittlement of Zirconium: An ab initio Investigation. Journal of Nu-clear Materials, 467, 311-319. https://doi.org/10.1016/j.jnucmat.2015.09.056
- 10. Maxwell, C.I., Torres, E. and Pencer, J. (2018) Molecular Dy-namics Study of Hydrogen-Vacancy Interactions in α- Zirconium. Journal of Nuclear Materials, 511, 341-352. https://doi.org/10.1016/j.jnucmat.2018.09.012
- 11. Christensen, M., Wolf, W., Freeman, C., et al. (2015) Diffu-sion of Point Defects, Nucleation of Dislocation Loops, and Effect of Hydrogen in hcp-Zr: Ab Initio and Classical Simu-lations. Journal of Nuclear Materials, 460, 82-96. https://doi.org/10.1016/j.jnucmat.2015.02.013
- 12. Zhu, X.Y., Lin, D.Y., Fang, J., Gao, X.Y., Zhao, Y.F. and Song, H.F. (2018) Structure and Thermodynamic Properties of Zirconium Hydrides by Structure Search Method and First Principles Calculations. Computational Materials Science, 150, 77-85. https://doi.org/10.1016/j.commatsci.2018.03.066
- 13. Zhang, Y.F., Bai, X.M., Yu, J.G., Tonks, M.R., Noord-hoek, M.J. and Phillpot, S.R. (2016) Homogeneous Hydride Formation Path in α-Zr: Molecular Dynamics Simulations with the Charge-Optimized Many-Body Potential. Acta Materialia, 111, 357-365. https://doi.org/10.1016/j.actamat.2016.03.079
- 14. Liu, S.J., Shi, S.Q., Huang, H. and Woo, C.H. (2002) Intera-tomic Potentials and Atomistic Calculations of Some Metal Hydride Systems. Journal of Alloys and Compounds, 330-332, 64-69. https://doi.org/10.1016/S0925-8388(01)01451-7
- 15. Noordhoek, M.J., Liang, T., Chiang, T.W., Sinnott, S.B. and Phillpot, S.R. (2014) Mechanisms of Zr Surface Corrosion Determined via Molecular Dynamics Simu-lations with Charge-Optimized Many-Body (COMB) Potentials. Journal of Nuclear Materials, 452, 285-295. https://doi.org/10.1016/j.jnucmat.2014.05.023
- 16. Thuinet, L. and Besson, R. (2011) Ab Initio Study of Competi-tive Hydride Formation in Zirconium Alloys. Intermetallics, 20, 24-32. https://doi.org/10.1016/j.intermet.2011.08.005
- 17. Moelans, N., Blanpain, B. and Wollants, P. (2007) An Intro-duction to Phase-Field Modeling of Microstructure Evolution. Calphad, 32, 268-294. https://doi.org/10.1016/j.calphad.2007.11.003
- 18. Thuinet, L., Legris, A., Zhang, L.F. and Ambard, A. (2013) Mesoscale Modeling of Coherent Zirconium Hydride Precipitation under an Applied Stress. Journal of Nuclear Materi-als, 438, 32-40. https://doi.org/10.1016/j.jnucmat.2013.02.034
- 19. Thuinet, L., De Backer, A. and Legris, A. (2012) Phase-Field Modeling of Precipitate Evolution Dynamics in Elastically Inhomogeneous Low-Symmetry Systems: Application to Hy-dride Precipitation in Zr. Acta Materialia, 60, 5311-5321. https://doi.org/10.1016/j.actamat.2012.05.041
- 20. Ma, X.Q., Shi, S.Q., Woo, C.H. and Chen, L.Q. (2002) Simu-lation of γ-Hydride Precipitation in bi-Crystalline Zirconium under Uniformly Applied Load. Materials Science and En-gineering: A, 334, 6-10. https://doi.org/10.1016/S0921-5093(01)01770-1
- 21. Ma, X.Q., Shi, S.Q., Woo, C.H. and Chen, L.Q. (2002) Ef-fect of Applied Load on Nucleation and Growth of γ-Hy- drides in Zirconium. Computational Materials Science, 23, 283-290. https://doi.org/10.1016/S0927-0256(01)00226-9
- 22. Shi, S.Q. and Xiao, Z.H. (2015) A Quantitative Phase Field Model for Hydride Precipitation in Zirconium Alloys: Part I. Development of Quantitative Free Energy Functional. Journal of Nuclear Materials, 459, 323-329. https://doi.org/10.1016/j.jnucmat.2014.03.013
- 23. Xiao, Z.H., Hao, M.J., Guo, X.H., Tang, G.Y. and Shi, S.Q. (2015) A Quantitative Phase Field Model for Hydride Precipitation in Zirconium Alloys: Part II. Modeling of Tempera-ture Dependent Hydride Precipitation. Journal of Nuclear Materials, 459, 330-338. https://doi.org/10.1016/j.jnucmat.2014.12.110
- 24. Heo, T.W., Colas, K.B., Motta, A.T., et al. (2019) A Phase-Field Model for Hydride Formation in Polycrystalline Metals: Application to δ-Hydride in Zirconium Alloys. Acta Materialia, 181, 262-277. https://doi.org/10.1016/j.actamat.2019.09.047
- 25. Bailey, J.E. (1963) Electron Microscope Observations on the Precipitation of Zirconium Hydride in Zirconium. Acta Metallurgica, 11, 267-280. https://doi.org/10.1016/0001-6160(63)90182-2
NOTES
*通讯作者。