Journal of Advances in Physical Chemistry
Vol.05 No.04(2016), Article ID:18975,10
pages
10.12677/JAPC.2016.54013
Spin Forbidden Reaction Mechanism of Th Activation of C2H4 in Gas Phase
Cuilan Wang
Jishishan County Jishi High School, Jishishan Gansu

Received: Oct. 30th, 2016; accepted: Nov. 15th, 2016; published: Nov. 18th, 2016
Copyright © 2016 by author and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Using UB3LYP method in density functional theory, the micro mechanism of the singlet and triplet potential energy surfaces of the three spin forbidden reactions Th(6d27S2) + C2H4→ThH3-CCH was investigated. Two reaction channels were found, and the two state reaction (TSR) was analyzed. Then we used the method of single point vertical excitation calculation of intrinsic coordinates Hammond and Yoshizawa’s hypothesis, found a series of potential energy surface crossing point of [crossing points (CPs)], and made the corresponding discussion. By calculation, the crossover of potential energy surface and the spin flip process are discussed. Using Harvey and other methods to optimize the minimum energy crossover (MECP), the possibility of the “inter system channeling” (ISC) between different potential energy surfaces in the reaction of Th and C2H4 is further discussed. In the end, we make a brief discussion on the frontier molecular orbital MECP2, which can be used as a guide for further theoretical research and experiment.
Keywords:Th Actives C2H4, Density Functional Theory (DFT), Potential Energy Surfaces, Minimum Energy Crossing Point (MECP)

气相中Th活化C2H4的自旋禁阻反应机理
王翠兰
积石山县积石中学,甘肃 积石山
Email: cuilan667608@126.com
收稿日期:2016年10月30日;录用日期:2016年11月15日;发布日期:2016年11月18日

摘 要
用密度泛函理论中的UB3LYP方法,研究了单重态和三重态势能面自旋禁阻反应Th(6d27S2) + C2H4→ThH3-CCH的微观机理。找到了两条反应通道,对其中涉及的两态反应(TSR)进行了分析。进而运用Hammond假设和Yoshizawa等的内禀坐标单点垂直激发计算的方法找出了一系列势能面交叉点[crossing points (CPs)],并作了相应的讨论。通过计算,讨论了势能面交叉和可能的自旋翻转过程。用Harvey等的方法优化出最低能量交叉(MECP),进一步讨论了Th与C2H4的反应中不同势能面之间的“系间窜越”(ISC)的可能性。最后,对前线分子轨道MECP2做了简单的讨论,这些理论结果可以作为进一步理论研究和实验的指导。
关键词 :Th活化C2H4,密度泛函理论(DFT),势能面,最低能量交叉点(MECP)

1. 引言
由于C-H键的活化在催化过程中起着决定性的作用,所以在实验和理论化学中引起化学家极大的兴趣。气相研究为许多合成反应和催化过程在分子水平上已经提供了洞悉机理的基本步骤 [1] [2] 。紧接着重排反应为许多合成反应和合成过程产生相关的中间物种展开了可能性 [3] 。C-H(X)的插入和随后烷烃和卤代烷的重排以及烯烃和许多过渡金属包括锕系已经被深入研究了 [4] - [11] 。特别是乙烯被激光熔融的过渡金属消除氢气的反应机理在近几年被广泛的关注 [12] [13] [14] [15] [16] 。同时,反应路径的细节也是理论研究的主体。Siegbahn, Blomberg, 和Svensson理论上调查了第二过渡金属原子活化乙烯中的C-H键,最后这个C-H键断裂导致H2的消除 [17] [18] 。他们的研究透露了过渡金属在乙烯复合物中首先形成一个强的键,这个乙烯复合物重新排列形成一个插入产品。这些研究者提出了消除H2的路径:M + ethylene(乙烯)→π complex (π 络合物)→insertion product(插入产品)→dihydrido complex (二氢复合物)→M-C2H2 + H2。
显然,在一系列动力学反应研究中,乙烯与过渡金属阳离子和中性原子的反应已经被祥细的做了研究 [19] - [27] 。相比之下,锕系的过渡金属原子较少的被广泛探索,尽管实验和理论正在扩展特别是锕系中性原子Th和U [28] [29] 。Andrews团队报道了运用激光熔融的方法对Th与U金属与乙烯反应的实验研究 [11] 。并且在B3LYP水平上对相关产物的结构、能量、频率等做了理论计算,来支撑他们的最低能量产物,但是机理问题未做详细的阐述。在目前的研究中,我们报道了锕系的过渡金属Th原子活化乙烯详细的势能面。为了更好的理解反应中自旋翻转过程,我们使用密度泛函理论对锕系的过渡金属Th原子活化乙烯中C-H键过程中涉及的两个势能面交叉点进行了详细的研究。对于钍与乙烯在气相中的反应,我们的计算产生了一个普遍的机理。
从理论的观点出发,通过Th与乙烯的反应调查显示了这个反应的最后产品是意想不到的三氢乙炔基复合物(ThH3-CCH)。值得注意的是,就Th与乙烯反应的这个路径中,氢气的消除反应我们也做了计算,但结果是计算失败,这就表明氢气的消除反应很缓慢,这点与实验观察很好的吻合。
2. 计算方法
Th与乙烯反应的三重态和单重态势能面做了详细的介绍。所有的计算采用Gaussian 03程序包 [30] 完成。我们用密度泛函(DFT)中的B3LYP方法 [31] [32] [33] ,非金属原子C、H [34] 分别采用6-311++G (d, p)、6-311++G (2df, 2pd)、6-311++G (3df, 3pd)基组,金属原子采用包含相对论效应核势能(relativistic effective core potential)的Stuttgart的赝势基组,对各个不同自旋态反应势能面上的反应物、中间体、过渡态和产物的构型进行了全参数优化。我们通过对每一个鞍点进行内禀反应坐标(IRC)计算,为的是在于跟踪从过渡态到相关最低点的最低能量反应路径,证实正确合适的过渡态是否被确定。就能量的二阶导数矩阵元来说,某一个势能面上的最低点没有负的本征值,鞍点处仅有一个负的本征值。为获得更精确的相对能量值,在B3LYP优化的几何构型基础上进一步采用CCSD(T) [35] 的方法,结合6-311++G (d, p)、6-311++G (3df, 3pd)基组对势能面上的各驻点进行了单点能计算。 我们对反应体系的两个反应势能面上的所有反应物、产物、中间体和过渡态的构型进行了全参数优化。为了更深入地探讨反应的微观机理,我们还运用NBO5.0 [36] 程序对自然键轨道做了NBO分析。
为了估计两个反应势能面交叉点(crossing point, CP),运用了Yoshizawa等 [37] 的内禀反应坐标单点垂直激发计算的方法,即在一自旋态的IRC反应坐标各点所对应的构型上,计算该构型在另一自旋态下的单点能量,得到了一自旋态势能面上IRC反应坐标投影在另一自旋态势能面上的势能曲线(但不是另一自旋态的IRC曲线),从而找到一自旋态IRC曲线与另一自旋态势能面相交的交叉点。再以CP点构型为依据,运用Harvey等 [38] 的方法优化得到最低能量交叉点(MECP)。
3. 结果与讨论
这部分概括了钍金属与乙烯反应的机理及各种物质的结构和能量参数。在不同自旋态下,各反应物、中间体、过渡态及产物的主要结构参数见图1以及反应路径的势能面见图2。在DFT和CCSD(T)的水平下采用不同基组所得到的能量分别列于表1和表2。从表1和表2可以看到CCSD水平下的能量无论从三重态还是单重态都比DFT下的能量高,所以我们就再没讨论CCSD水平下的能量。以下的能量讨论都指的是B3LYP/6-311++G (3df, 3pd)高机组水平下的能量。

Table 1. At the level of DFT-B3LYP Th and C2H4 reaction of each point energy
表1. 在DFT-B3LYP水平下Th与C2H4反应的各驻点能量
aEZPE: zero-point vibrational energy (a.u.). bET: total energy (a.u.). cER: relative energy with correction of the zero-point energy (kcal/mol)



 1IM0(A1, C2v) 1TS01(A, C1) 1IM1(A″, Cs) 1TS12(A, C1)
1IM0(A1, C2v) 1TS01(A, C1) 1IM1(A″, Cs) 1TS12(A, C1)


 1IM2(A′, Cs) 1TS23(A′, C1) 1TS24(A′, Cs)
1IM2(A′, Cs) 1TS23(A′, C1) 1TS24(A′, Cs)


 1IM3(A′, Cs) 1TS34(A, C1) 1IM4(A′, Cs)
1IM3(A′, Cs) 1TS34(A, C1) 1IM4(A′, Cs)



 3IM0(A1, C2v) 3TS01(A, C1) 3IM1(A″, Cs) 3TS12(A, C1)
3IM0(A1, C2v) 3TS01(A, C1) 3IM1(A″, Cs) 3TS12(A, C1)


 3IM2(A′, Cs) 3TS23(A, C1) 3TS24(A, C1)
3IM2(A′, Cs) 3TS23(A, C1) 3TS24(A, C1)

 3IM3(A1, C2v) 3TS34(A′, Cs) 3IM4(A′, Cs)
3IM3(A1, C2v) 3TS34(A′, Cs) 3IM4(A′, Cs)
Figure 1. UB3LYP/6-311++G (3df, 3pd) the geometry level under different spin state potential energy surface of stationary points
图1. UB3LYP/6-311++G (3df, 3pd)水平下不同自旋态势能面上各驻点的几何构型

Figure 2. Path diagram of C2H4 reaction with Th on two potential energy surfaces
图2. Th在两个势能面上与C2H4反应的路径示意图
Table 2. The single point energy of the reaction of Th + C2H4 at the CCSD level
表2. 在CCSD水平下Th + C2H4的反应的单点能量
3.1. 三重态势能面
Th (6d27S2, 3F) 基态的能量比激发态低13.4 kcal/mol的能量,因此Th与乙烯在低能量状况下更有利于反应。对于三重态势能面,我们可以看到总的反应是一个插入机理,首先,第一步基态3Th 与乙烯分子相互碰撞形成3IM0;第二步3IM0经过过渡态3TS01,克服29.6 kcal/mol的能垒相应的一个H原子从碳原子转移到金属Th原子上,形成插入化合物ThH-C2H3 (3IM1),该过程是放热反应,放热仅仅为1.9 kcal/mol的能量。接下来反应继续进行,3IM1通过第二个氢转移形成二氢复合物C2H2=ThH2 (3IM2),翻越一个活化能垒24.0 kcal/mol,同时吸收5.1 kcal/mol的能量。我们成功地计算了3IM2的结构,它是三中心原子复合物,并且这个过程的过渡态与高自旋有关系。3IM2非常有趣,因为从3IM2开始有两条路径。第一条路径是3IM2通过过渡态3TS23产生亚乙烯基复合物ThH2=C=CH2 (3IM3)。接着活化第三个C-H键通过一个很重要的过渡态3TS34形成三氢乙炔基复合物ThH3-CCH (3IM4)。这一步吸收热量10.0 kcal/mol,并且爬跃40.3 kcal/mol的能垒。如果沿着第二条路径反应,3IM2通过过渡态3TS24直接形成3IM4。我们定位了3TS24的一个鞍点从3IM2和3IM4中。这个过渡态3TS24仅有一个虚频269.79i cm−1,其中能垒是52.6 kcal/mol。分析三重态势能面,我们可以看到第二个H和第三个H转移是吸热过程。
3.2. 单重态势能面
让我们回到单重态势能面上。首先是一个分子加合物的形成由于Th与乙烯的碰撞反应。与三重态相比较,这个单重态也就是激发态Th与乙烯反应能量上比三重态高出13.5 kcal/mol反应开始于1IM0,从它转移第一个氢原子经过过渡态1TS01克服活化能垒24.4 kcal/mol形成插入中间体1IM1。接着从1IM1上转移第二个氢原子生成CH2=ThH2 (1IM2),这个过程有一个活化能垒13.4 kcal/mol。1IM2以后的反应相似于三重态,单重态也有两条路径。第一条路径的反应是通过爬跃一个活化能垒24.1 kcal/mol形成ThH2=C=CH2 (1IM3)。最后第三个氢转移导致一个可能的产物三氢乙炔基复合物ThH3-CCH(1IM4)的生成。这个过程是放热的,15.6 kcal/mol,并且有一个能垒比较高的过渡态1TS34,能垒为62.1 kcal/mol,这个过程看作是高能垒机理。这个路径总的反应放热73.1 kcal/mol。对于第二条路径,我们的计算显示了1IM2不得不克服过渡态1TS24的活化能垒46.9 kcal/mol才能生成1IM4。从1IM2到1IM4的这个过程仅仅吸热4.6 kcal/mol。中间体1IM3中,Th-C键的距离为2.153 Å,同1IM4相比Th-C键的距离为2.412 Å。1IM3中C-H键的距离2.090 Å而1IM4中C-H键的距离2.075 Å。目前的计算表明,最积极良好的反应路径是产生三氢乙炔基复合物ThH3-CCH,这就支持了实验观察。我们计算的反应路径与Andrews等人的实验和理论是非常一致的。
3.3. 势能面间的交叉
从图2可以看出,在第一条路径上势能面有交叉现象。这个渠道是自旋禁阻的,是三重态和单重态之间的交叉。我们推测了交叉点位于两个可能区域的两个势能面间。一个可能的区域是Th刚开始与乙烯相互作用的过程中,这个区域位于1IM0 和3IM0之前。从图2可以看到1IM0在能量上比3IM0低,它们的结构是相似的。为了找出这个交叉点,势能面的扫描是可行的办法。另为的一个可能区域位于IM3和IM4之间,1TS34在能量上比3TS34高,1IM3和1IM4的能量比3IM3和3IM4的都低。为了确认第二个可能区域,我们选择了Yoshizawa等人的一个简单方法来确定两个不同势能面间的交叉点。这主要的过程是执行一系列的单点能计算沿着IRC在另一自旋态下,反之亦然 [39] 。从亚乙烯基化合物ThH2=C=CH2 (1IM3)开始,沿着1IM3→3TS34→1IM4的IRC,在B3LYP/6-311++G (3df, 3pd)水平下,在单重态下执行了一系列单点能计算,我们计算得到了单重态势能面下的单点能。我们发现单重态势能面总是低于三重态势能面,因此我们推断交叉点就在这个区域。图3显示了三重态和单重态势能面间的最低能量交叉点的结构图。值得注意的是,交叉点MECP1对降低活化能垒起着非常重要的作用,这是因为MECP1位于反应通道的入口处,在反应的过程中起着重要的角色。为了更深入的了解自旋翻转和势能面之间的交叉现象,我们对MECP2点进行了NBO轨道分析和前线分子轨道相互作用分析,获得了些有用的信息。如图4所示,a是Th原子的dz2轨道,是三重态的最高单占轨道即HOMO,同时也是单重态的最低空轨道即LUMO。b是Th原子的dxz轨道,是三重态的最低单占轨道即SOMO,同时是单重态的最高双占轨道。在这个例子中,a轨道上的α电子会自旋翻转翻转,也就是说,单电子从过渡金属Nb的a轨道(dz2)跳跃到b轨道(dxz)上,完成由三重态向单重态的转变。因为在一个原子的不同轨道上单电子的转移是允许的 [40] ,所以系间窜越是有效的。对于最低能量交叉点MECP3,在整个反应路径中没有起到重要的作用,因此,这个交叉点在这儿不再仔细的考虑。

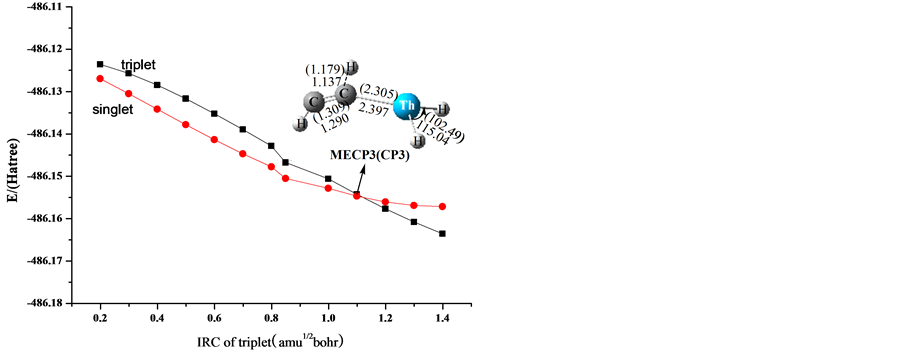
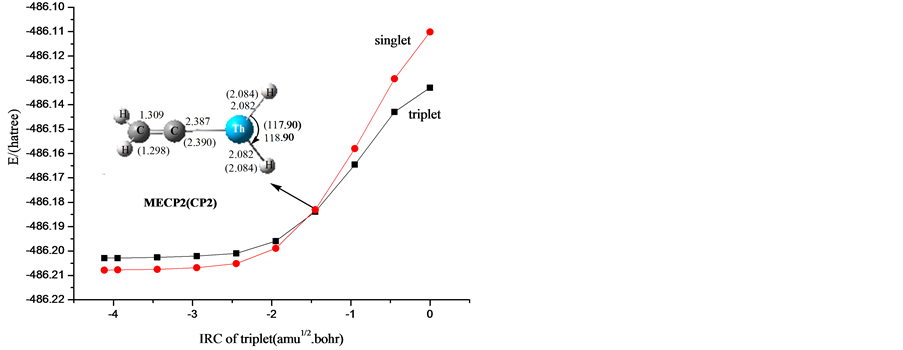
Figure 3. Schematic diagram of the structure of the lowest energy crossing points along the potential energy
图3. 沿着势能面的各个最低能量交叉点的结构示意图

Figure 4. Analysis of the frontier molecular orbital at MECP2
图4. MECP2处的前线分子轨道分析
3.4. 气相反应机理
沿着三重态和单重态反应路径,金属原子Th与乙烯的相互作用被描述为Chatt-Dewar-Duncanson机理 [41] 。从图1中的IM0结构可以看到,刚开始金属Th原子与乙烯相互碰撞,乙烯提供部分π电子给金属的σ空轨道。结果,乙烯的π键被消弱了,这个π*轨道在能量上降低了,电子被金属原子d轨道上的反馈键所接受,这样就形成了π键,这样就形成了一个重金属环丙烷的复合物。相对于自然的乙烯分子,IM0这个复合物的C-C键被拉长了,C-C-H键角被扭曲了。另为Th-C键的长度是比较短的,这就足以说明有两个共价键形成了。接下来的反应是Th插入到乙烯的C-H键中,通过一个过渡态TS01,沿着高自旋和低自旋的势能面,转移一个H原子从碳原子到金属原子上,生成中间体H-Th-C2H3。我们看到这两个过渡态的能垒是不同的,单重态位于三重态的下面。由于低自旋下两个金属电子参与到两个共价键的结构当中,所以插入复合物H-Th-C2H3比较稳定。因此这个反应沿着单重态路径继续进行,通过过渡态TS12转移一个β氢原子生成最稳定的复合物ThH2-C2H2(IM2)。对于IM2中间体,非常有趣。这个中间体比较起初的自由金属能够改变状态的相对稳定性。例如中间体3IM2,值得注意的是C-C键和C-Th键是最短的。这个金属原子在高氧化态下趋于形成一个强健 [42] 。与此同时反馈增强,预期π*轨道出现在3IM2中,进一步加强了C-M键。从NBO的C-C π*的占有轨道上我们看到,3IM2比3IM1低,分别是0.0144和0.1780。这就明显的导致了较短的C-C键。相反的,二氢复合物(ThH2-C2H2) NBO的C-Th π*的占有轨道,比插入中间体(ThH-C2H2)的高,分别是0.0362和0.0115。很明显,为了进一步研究的需要,我们留下了更多有趣的细节,为的是将来做研究。经过IM2后,有两条路径生成最终产物。第一条路径是IM2通过过渡态TS23形成亚乙烯基复合物IM3。接着活化第三个C-H键通过一个重要的过渡态TS34最后产生三氢乙炔基复合物IM4。第二条路径是IM2克服过渡态TS24直接形成最终产物IM4。有图2通过比较我们看到,无论是三重态还是单重态都是第一条路径有利。王永成等人 [43] 已经做了一个第五过渡金属与乙烯反应的相似计算。
4. 结论
采用密度泛函UB3LYP/6-311++G (d, p)//6-311++G (2df, 2pd)//6-311++G (3df, 3pd)方法对气相反应Th+H2→ThH3-CCH的微观机理进行了计算研究,得出了以下结论。
1) 本文在单重态和三重态两个势能面上探究了此反应。Th (1D)和Th(3F)与乙烯都有相似的反应机理,它们都是一个典型的插入机理。
2) 最小能量反应路径并不是两个势能面的其中一个,Th与乙烯的最小能量反应路径描述如下:
Th(3F) + C2H4(Ag)→MECP1→1IM0→1TS01→1IM1→1TS12→1IM2→1TS23→1IM3→MECP2→3TS34→ MECP3→1IM4。
3) 反应过程中发生了三重态和单重态两个势能面交叉,交叉点CP1、CP2和CP3分别出现在入口处、1IM3→3TS34和3TS34→1IM4的路径上,使反应始终保持在能量较低的是能面上。
4) 目前的工作第一次做了一个详细的研究,为更好地理解锕系其它过渡金属原子与简单碳氢化合物的反应起了一点帮助。
致谢
我们感谢国家自然基金(Grant No. 20873102)同时感谢省计算中心提供的计算。
文章引用
王翠兰. 气相中Th活化C2H4的自旋禁阻反应机理
Spin Forbidden Reaction Mechanism of Th Activation of C2H4 in Gas Phase[J]. 物理化学进展, 2016, 05(04): 112-121. http://dx.doi.org/10.12677/JAPC.2016.54013
参考文献 (References)
- 1. Schröder, D., Shaik, S. and Schwarz, H. (2000) Two-State Reactivity as a New Concept in Organometallic Chemistry. Accounts of Chemical Research, 33, 139-145. http://dx.doi.org/10.1021/ar990028j
- 2. Ogliaro, F., Harris, N., Cohen, S., Filatov, M., de Visser, S.P. and Shaik, S. (2000) A Model “Rebound” Mechanism of Hydroxylation by Cy-tochrome P450: Stepwise and Effectively Concerted Pathways, and Their Reactivity Patterns. Journal of the American Chemical Society, 122, 8977-8989. http://dx.doi.org/10.1021/ja991878x
- 3. Olson, D.E. and Du Bois, J. (2008) Catalytic C-H Amination for the Preparation of Substituted 1,2-Diamines. Journal of the American Chemical Society, 130, 11248-11249. http://dx.doi.org/10.1021/ja803344v
- 4. Andrews, L. and Cho, H.G. (2006) Matrix Preparation and Spectroscopic and Theoretical Investigations of Simple Methylidene and Methylidyne Complexes of Group 4-6 Transition Metals. Organometallics, 25, 4040-4053. http://dx.doi.org/10.1021/om060318l
- 5. Davies, H.M.L. and Beckwith, R.E.J. (2003) Catalytic Enantioselective C-H Activation by Means of Metal-Carbenoid- Induced C-H Insertion. Chemical Reviews, 103, 2861-2904. http://dx.doi.org/10.1021/cr0200217
- 6. Proctor, D.L. and Davis, H.F. (2008) Vibrational vs. Translational Energy in Promoting a Prototype Metal-Hydrocar- bon Insertion Reaction. Proceedings of the National Academy of Sciences of the United States of America, 105, 12673- 12677. http://dx.doi.org/10.1073/pnas.0801170105
- 7. Lee, Y.K., Manceron, L. and Papai, I. (1997) An IR Matrix Isolation and DFT Theoretical Study of the First Steps of the Ti (0) Ethylene Reaction: Vinyl Titanium Hydride and Titanacyclopropene. Journal of Physical Chemistry A, 101, 9650-9659. http://dx.doi.org/10.1021/jp971870e
- 8. Cho, H.G. and Andrews, L. (2004) Hydrogen Elimination from Ethylene by Laser-Ablated Zr Atoms: An Infrared Spectroscopic Investigation of the Reaction Intermediates in a Solid Argon Matrix. Journal of Physical Chemistry A, 108, 3965-3972. http://dx.doi.org/10.1021/jp049566q
- 9. Cho, H.G. and Andrews, L. (2007) Matrix Infrared Spectroscopic Studies of the MH-C2H3 and MH2-C2H2 Intermediates in the Reactions of Ethylene with Laser-Ablated Group 5 Metal Atoms. Journal of Physical Chemistry A, 111, 5201-5210. http://dx.doi.org/10.1021/jp0702806
- 10. Cho, H.G. and Andrews, L. (2008) Infrared Spectra of Metallacyclo-propane, Insertion, and Dihydrido Complex Products in Reactions of Laser-Ablated Group 6 Metal Atoms with Ethylene Molecules. Journal of Physical Chemistry A, 112, 12071-12081. http://dx.doi.org/10.1021/jp806110h
- 11. Cho, H.G. and Andrews, L. (2009) Matrix Infrared Spectra of Dihydrido Cyclic and Trihydrido Ethynyl Products from Reactions of Th and U Atoms with Ethylene Molecules. Journal of Physical Chemistry A, 113, 5073-5081. http://dx.doi.org/10.1021/jp900610c
- 12. Yi, S.S., Blomberg, M.R.A., Siegbahn, P.E.M. and Weisshaar, J.C. (1998) Statistical Modeling of Gas-Phase Organometallic Reactions Based on Density Functional Theory: Ni+ + C3H8. Journal of Physical Chemistry A, 102, 395-411. http://dx.doi.org/10.1021/jp972674a
- 13. Blomberg, M., Siegbahn, P.E.M., Yi, S.S., Noll, R.J. and Weisshaar, J.C. (1999) Gas-Phase Ni+(2D5/2) + n-C4H10 Reaction Dynamics in Real Time: Experiment and Statistical Modeling Based on Density Functional Theory. Journal of Physical Chemistry A, 103, 7254-7267. http://dx.doi.org/10.1021/jp991561j
- 14. Jiao, C.Q. and Freiser, B.S. (1995) Reactions of Nbn+ (n = 2 - 6) with Ethylene in the Gas Phase: Collision-Induced Dissociation Studies of Ionic Products. Journal of Physical Chemistry A, 99, 3969-3977. http://dx.doi.org/10.1021/j100012a017
- 15. Stauffer, H.U., Hinrichs, R.Z., Schroden, J.J. and Davis, H.F. (2000) Dynamics of H2 and C2H4 Elimination in the Y + C2H6 Reaction. Journal of Physical Chemistry A, 104, 1107-1116. http://dx.doi.org/10.1021/jp993525q
- 16. Parnis, J.M.P., Lafleur, R.D. and Rayner, D.M. (1995) Hydrocarbon Reactivity with Early Transition Metal Atoms and Neutral Diatomic Metal Oxides in the Gas Phase. Journal of Physical Chemistry A, 99, 673-680. http://dx.doi.org/10.1021/j100002a035
- 17. Siegbahn, P.E.M., Blomberg, M.R.A. and Svensson, M. (1993) A Theoretical Study of the Activation of the Carbon- Hydrogen Bond in Ethylene by Second-Row Transition-Metal Atoms. Journal of the American Chemical Society, 115, 1952-1958. http://dx.doi.org/10.1021/ja00058a048
- 18. Blomberg, M.R.A., Siegbahn, P.E.M. and Svensson, M. (1992) Theoretical Study of the Binding of Ethylene to Second-Row Transition Metal Atoms. Journal of Physical Chemistry A, 96, 9794-9800. http://dx.doi.org/10.1021/j100203a040
- 19. Thompson, M.G.K. and Parnis, J.M. (2005) Photoinduced Ethane Formation from Reaction of Ethene with Matrix- Isolated Ti, V, or Nb Atoms. Journal of Physical Chemistry A, 109, 9465-9470. http://dx.doi.org/10.1021/jp0447542
- 20. Reichert, E.L., Yi, S.S. and Weisshaar, J.C. (2000) Bimo-lecular Ion-Molecule Collisions in Real Time: Co+ (3f4) + n-Butane and Isobutane Reactions. International Journal of Mass Spectrometry, 196, 55-69. http://dx.doi.org/10.1016/S1387-3806(99)00193-1
- 21. Carroll, J.J., Haug, K.L., Weisshaar, J.C., Blomberg, M.R.A., Siegbahn, P.E.M. and Svensson, M. (1995) Gas Phase Reactions of Second-Row Transition Metal Atoms with Small Hydrocarbons: Experiment and Theory. The Journal of Physical Chemistry, 99, 13955-13969. http://dx.doi.org/10.1021/j100038a030
- 22. Willis, P.A., Stauffer, H.U., Hinrichs, R.Z. and Davis, H.F. (1999) Reaction Dynamics of Zr and Nb with Ethylene. The Journal of Physical Chemistry A, 103, 3706-3720. http://dx.doi.org/10.1021/jp9846633
- 23. Gidden, J., van Koppen, P.A.M. and Bowers, M.T. (1997) Dehydroge-nation of Ethene by Ti+ and V+: Excited State Effects on the Mechanism for C-H Bond Activation from Kinetic Energy Release Distributions. Journal of the American Chemical Society, 119, 3935-3941. http://dx.doi.org/10.1021/ja964377+
- 24. Guo, B.C. and Castleman, Jr., A.W. (1992) Dehydrogenation of Ethylene and Propylene and Ethylene Polymerization Induced by Titanium(1+) in the Gas Phase. Journal of the American Chemical Society, 114, 6152-6158. http://dx.doi.org/10.1021/ja00041a037
- 25. Sanders, L., Hanton, S. and Weisshaar, J.C. (1987) Electron Spin State Selectivity in Transition Metal Ion Reactions: V+(a3F) + C2H6→VC2H4+ + H2. The Journal of Physical Chemistry, 91, 5145-5148. http://dx.doi.org/10.1021/j100304a001
- 26. Sievers, M.R., Jarvis, L.M. and Armentrout, P.B. (1998) Transi-tion-Metal Ethene Bonds: Thermochemistry of M+(C2H4)n (M = Ti-Cu, n = 1 and 2) Complexes. Journal of the American Chemical Society, 120, 1891-1899. http://dx.doi.org/10.1021/ja973834z
- 27. Simon, A., MacAleese, L., Boissel, P. and Maitre, P. (2002) Towards the Characterization of the Mechanism of the Sequential Activation of Four Methane Molecules by Ta+. International Journal of Mass Spectrometry, 219, 457-473. http://dx.doi.org/10.1016/S1387-3806(02)00700-5
- 28. Cho, H.G. and Andrews, L. (2005) Infrared Spectrum and Structure of CH2=ThH2. The Journal of Physical Chemistry A, 109, 6796-6798. http://dx.doi.org/10.1021/jp052918o
- 29. Lyon, J.T., Andrews, L., et al. (2007) Infrared Spectrum and Bonding in Uranium Methylidene Dihydride, CH2=UH2. Inorganic Chemistry, 46, 4917-4925. http://dx.doi.org/10.1021/ic062407w
- 30. Frisch, M.J., et al. (2003) GAUSSIAN 03 (Revision-E.01), Gaussian, Inc., Pittsburgh, PA.
- 31. Liu, Z., Zhong, L., Yang, Y., Cheng, R. and Liu, B. (2011) DFT and CASPT2 Study on the Mechanism of Ethylene Dimerization over Cr(II)OH+ Cation. The Journal of Physical Chemistry A, 115, 8131-8141. http://dx.doi.org/10.1021/jp111108p
- 32. Wang, T., Brudvig, G. and Batista, V.S. (2010) Characterization of Proton Coupled Electron Transfer in a Biomimetic Oxomanganese Complex: Evaluation of the DFT B3LYP Level of Theory. Journal of Chemical Theory and Computation, 6, 755-760. http://dx.doi.org/10.1021/ct900615b
- 33. Cao, Z. and Ren, T. (2011) DFT Study of Electronic Properties of 3d Metal Complexes of σ-Geminal Diethynylethenes (gem-DEEs). Organometallics, 30, 245-250. http://dx.doi.org/10.1021/om100870k
- 34. McLean, A.D. and Chandler, G.S. (1980) Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z=11-18. The Journal of Chemical Physics, 72, 5639-5648. http://dx.doi.org/10.1063/1.438980
- 35. Raghavachari, K., Trucks, G.W. and Pople, J.A. (1989) A Fifth-Order Perturbation Comparison of Electron Correlation Theories. Chemical Physics Letters, 157, 479-483. http://dx.doi.org/10.1016/S0009-2614(89)87395-6
- 36. Glendening, E.D., Badenhoop, J.K., Reed, A.E., Carpenter, J.E., Bohmann, J.A., Morales, C.M. and Weinhold, F. (2001) NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin, Madison.
- 37. Yoshizawa, K., Shiota, Y. and Yamabe, T. (1999) Methane-Methanol Conversion by MnO+, FeO+, and CoO+: A Theoretical Study of Catalytic Selectivity. Journal of the American Chemical Society, 120, 564-572. http://dx.doi.org/10.1021/ja971723u
- 38. Harvey, J.N., Aschi, M., Schwarz, H. and Koch, W. (1998) The Singlet and Triplet States of Phenyl Cation. A Hybrid Approach for Locating Minimum Energy Crossing Points between Non-Interacting Potential Energy Surfaces. Theoretical Chemistry Accounts, 99, 95-99. http://dx.doi.org/10.1007/s002140050309
- 39. Dai, G.L. and Fan, K.N. (2006) Theoretical Study of the Reaction of Sc+ with SCO in Gas Phase. Journal of Molecular Structure: THEOCHEM, 778, 55-61. http://dx.doi.org/10.1016/j.theochem.2006.08.043
- 40. Turro, N.J. (1987) Modern Molecular Photochemistry. Science Press, Beijing.
- 41. Chatt, J. and Duncanson, L.A. (1953) 586. Olefin Co-Ordination Compounds. Part III. In-fra-Red Spectra and Structure: Attempted Preparation of Acetylene Complexes. Journal of the Chemical Society, 2939-2947. http://dx.doi.org/10.1039/jr9530002939
- 42. Wang, X. and Andrews, L. (2002) Neon Matrix Infrared Spectra and DFT Calculation of Tungsten Hydrides WHx (x = 1-4, 6). The Journal of Physical Chemistry A, 106, 6720-6729. http://dx.doi.org/10.1021/jp025920d
- 43. Wang, C.L., Wang, Y.C., Jin, Y.Z., Ji, D.F., La, M.J., Ma, W.P. and Nian, J.Y. (2011) Theoretical Study of the C-H Bond in C2H4 by the Group 5 Metal Actoms (V, Nb, Ta) in the Gas-Phase. Computational and Theoretical Chemistry, 974, 43-51. http://dx.doi.org/10.1016/j.comptc.2011.07.010