Journal of Organic Chemistry Research
Vol.05 No.02(2017), Article ID:20878,4
pages
10.12677/JOCR.2017.52013
The Synthesis and Quantum Chemistry Calculation Based on 2-Thiol Pyridine Ligand
Liansheng Zhang1, Wei Wan1, Cuijuan Wang1, Xiaoyang Xiong1, Xin Zhao2, Zexian Li1, Zhibin Chen1
1Dept. College of Life Science and Engineering, Southwest Jiaotong University, Chengdu Sichuan
2Dept. College of Mechanical Engineering, Southwest Jiaotong University, Chengdu Sichuan

Received: May 15th, 2017; accepted: Jun. 4th, 2017; published: Jun. 7th, 2017

ABSTRACT
A six-core cluster compound Cu6(C5H4NS)6, was prepared by layered a N, N-Dimethylformamide solution of Cu(CH3COO)2·H2O on a dichloromethane solution of Dpds. The quantum chemistry calculation of the complex was carried out by using the B3LYP calculation method of density functional theory (DFT) and the NBO calculation method; at the same time, the research of frontier orbital is carried out. The results showed that the six-core copper (II) cluster complex contains six crystallographically unique Cu (II) ions and six 2-mercapto pyridine anions. It is found that the theoretical values are in agreement with the experimental ones, and the error is within the allowable range. And the completion of theoretical calculations proved the reliability of the experimental data.
Keywords:Dpds, Coordination Compounds, Functional Theory, B3LYP, NBO Calculations
2-巯基吡啶配合物的合成及量子化学计算
张连生1,万玮1,王萃娟1,熊霄阳1,赵鑫2,李泽贤1,陈志斌1
1西南交通大学生命科学与工程学院,四川 成都
2西南交通大学机械工程学院,四川 成都
收稿日期:2017年5月15日;录用日期:2017年6月4日;发布日期:2017年6月7日

摘 要
基于配体2,2’-二硫二吡啶(简称Dpds),采用分层扩散法,合成出六核簇状配位化合物Cu6(C5H4NS)6。采用密度泛函理论(DFT)下的B3LYP计算方法、NBO计算方法对该配合物进行量子化学计算,同时进行了前线轨道的研究。结果表明:每个配合物含六个铜离子、六个2-巯基吡啶阴离子,优化后的理论值与实验值比较吻合,误差在允许的范围内,理论计算的完成证明了实验数据的可靠性。
关键词 :2,2’-二硫二吡啶,配位化合物,密度泛函理论,B3LYP,NBO计算

Copyright © 2017 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
金属有机骨架化合物是一种非常重要的晶态材料,人们通过分子设计合成出大量结构新颖的金属有机框架化合物,研究了它们的拓扑结构,并对其在磁性、气体吸附与分离、选择性催化、非线性光学等领域内潜在的应用价值进行了探索 [1] [2] [3] 。其中含氮配体配合物是金属有机骨架化合物中非常重要的一类。因为含氮配体中的氮原子数目较多、氮原子与金属离子配位的能力强,所以配位反应比较容易发生,能够用于合成结构新颖的配位化合物。随着化学理论的发展及计算机性能的提高,集化学和计算于一体的计算化学,正在逐渐走向成熟。计算化学建立在理论的演绎上,通过求解一系列的方程,来解决化学上的问题。将计算化学的理论和方法应用到配合物的研究上,能够促进配位化学的发展。
本文采用2,2’-二硫二吡啶(简称Dpds)作为配体,它理论上能够与金属离子发生配位反应。但是从另一方面来说,二硫键容易断裂,含有二硫键的化合物很不稳定 [4] [5] 。实验中我们并没有合成出以二硫二吡啶为配体的配合物,而是得到了以巯基吡啶(二硫二吡啶的分解产物)为配体的六核铜簇合物。这种六核铜簇合物的合成已经有文献报导 [6] [7] ,但是本文中的合成方法与之前文献报导方法有很大的区别。文献 [6] 和 [7] 中使用的都是水热法,文献 [6] 中使用[Cu(PPh3)2(MeCN)2]ClO4和2-巯基-N-氧化吡啶,文献 [7] 中使用[Cu(MeCN)2]BF6和2-巯基吡啶。本文使用的是二硫二吡啶和乙酸铜,并且是在反应条件温和的试管中进行反应的。然后通过XRD单晶衍射 [8] [9] 对配合物进行结构测定。另外将对配合物进行量子化学计算和研究。
2. 实验部分
配合物Cu6(C5H4NS)6的合成
配合物的合成方法是:分层扩散法。称取0.05 mmol (11.0 mg) Dpds,溶于5 mL体积比为1:1的DMF-二氯甲烷混合溶剂,搅拌半小时,完全溶解后倒入试管。将5 mL体积比为2:1的DMF-二氯甲烷混合溶剂缓慢加入上述试管。称取0.05 mmol (10.0 mg)乙酸铜,溶于5 mL DMF,完全溶解后缓慢加入上述试管。试管中的三种溶液出现分层,将试管密封,室温静置。随着时间的推移,三种溶剂渐渐互溶,三周后得到黄色透明、呈长方体状的该配合物的晶体。以二硫二吡啶为标准,计算产率为31.2%。配合物Cu6(C5H4NS)6的元素分析测试值(%)为:C 34.39,H 2.24,N 18.13;计算值(%)为:C 34.48,H 2.30,N 18.05。
3. 结果与讨论
3.1. 基于配体Dpds的配位化合物的结构表征
3.1.1. 配合物Cu6(C5H4NS)6的晶体结构测定
配合物的晶体结构测定方法为:选取形状规则并且大小适合的单晶样品,常温下放在Bruker Smart-APEXII CCD衍射仪上,用经过石墨单色器单色化的Mo-Kα射线(λ = 0.071073 nm),以ω-f 方式扫描收集配合物的衍射数据。全部衍射强度数据通过Lp校正和吸收校正 [10] 。通过SHELXTL [11] 程序完成结构解析,然后用直接法将晶体结构解出。全部氢原子及非氢原子的坐标、及各向异性相关参数都由全矩阵最小二乘法F2精修,其中全部氢原子坐标由差值电子密度函数并结合几何分析方法获得;全部非氢原子则经Fourier合成及差值电子密度函数修正得到。所有相关计算均使用SHELXL-97程序包完成 [10] [11] 。配合物的晶体学数据已列于表1。
3.1.2. 配合物Cu6(C5H4NS)6的晶体结构描述
配合物的X-射线单晶衍射测试结果显示:配合物属于单斜晶系,P2(1)/n空间群。配合物每个分子含有六个铜离子、六个2-巯基吡啶阴离子。如图1所示,整个分子为1个分立的六核簇合物,六个2-巯基吡啶通过Cu-N、Cu-S配位键与Cu6核心结合。配合物的结构具有很高的对称性,每个S原子和两个相邻的铜原子配位,Cu-S键的键长在2.223~2.259 Å范围内。每个N原子和一个铜原子配位,Cu-N键的键长在2.031~2.037 Å范围内。
3.2. 配合物的量子化学计算
配合物和配体2-巯基吡啶离子的量子化学计算
配合物的结构为零维,不对称单元间没有配位键这种强相互作用力。我们运用Gaussian 03 [12] 程序,采用密度泛函理论下的B3LYP计算方法 [13] [14] ,对配合物以及配体2-巯基吡啶去质子状态下的2-巯基吡啶离子进行量子化学计算。对2-巯基吡啶离子的计算,我们使用6-31G(d)基组。对配合物的计算,我们使用混合基组,对C、H、S、N使用6-31G(d)基组,对Cu使用LANL2DZ赝势基组 [15] 。计算的内容包括:几何优化及在其基础上的频率计算,并分析了电荷布局和前线轨道。配合物键长、键角的计算值和理论值列于表2。比较结果发现,优化后的理论值与实验值比较吻合,误差在允许的范围内。键长的理论值比实验值偏长,其中Cu1-Cu3键的理论值比实验值大0.183 Å,这可以解释为在B3LYP方法优化下,键长普遍偏长。配合物和配体2-巯基吡啶离子的Mulliken [16] [17] 电荷列于表3。由表中数据可知,配合物中铜原子的电荷由1 (在配合物中铜离子的价态是+1)降到了0.725,这说明铜原子和配体发生了配位作用 [18] [19] 。与自由配体2-巯基吡啶离子相比,配合物中N1、S1的电荷增加,这些结果也向我们展示了铜原子和配体之间的配位作用。
通过NBO计算,我们得到了配合物和配体2-巯基吡啶离子的中各原子的电荷布局和自然电荷的信息,部分原子的信息列于表4。由表中数据可知,与配体2-巯基吡啶离子相比,在配合物中,硫原子的电荷升高。这是因为2-巯基吡啶阴离子的硫原子上积聚着负电荷,与带正电荷的铜原子配位后,电子流向铜原子,从而硫原子自身的电荷升高。在配合物中,氮原子外层轨道中的电子数量增加,氮原子的自然电荷略微降低。这可以解释为:配位键作用增加了氮原子周围的电子云密度 [18] [19] 。因为铜离子是吸电子基团,在这种吸引力作用下,Dpds配体中更多的电子集中在参与配位的氮原子的附近。
图2展示了配体2-巯基吡啶离子的前线轨道。在配体的HOMO(最高占据轨道)轨道中,硫原子和氮原子占到了很大的比例。在我们的实验中,配体中的硫原子、氮原子发生配位作用,这也验证了计算结果。图3展示了配合物的HOMO轨道的三维结构,正如我们预料的那样,配体2-巯基吡啶离子和铜原子的电子云发生了重叠,证明了配位键的存在。

Table 1. Crystallographic Data
表1. 晶体学数据
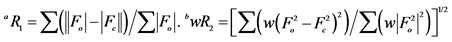
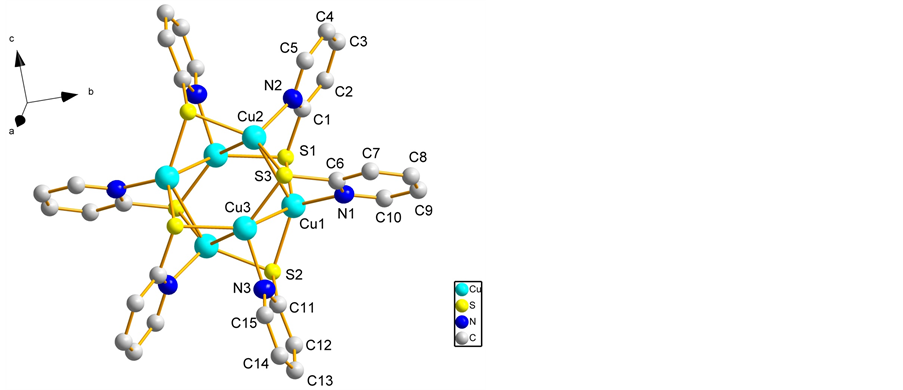
Figure 1. The coordination environment of Cu6(C5H4NS)6
图1. Cu6(C5H4NS)6的配位环境示意图
Table 2. Complex bond lengths (Å) and angles (˚) the theoretical and experimental values
表2. 配合物键长(Å)和键角(˚)的理论值与实验值
Table 3. The Mulliken charge of Dpds ion and complex
表3. 配合物和配体2-巯基吡啶离子的Mulliken电荷(e)
Table 4. The charge distribution and natural charge for complex and Dpds
表4. 配合物和配体Dpds的电荷布局(e)和自然电荷(e)
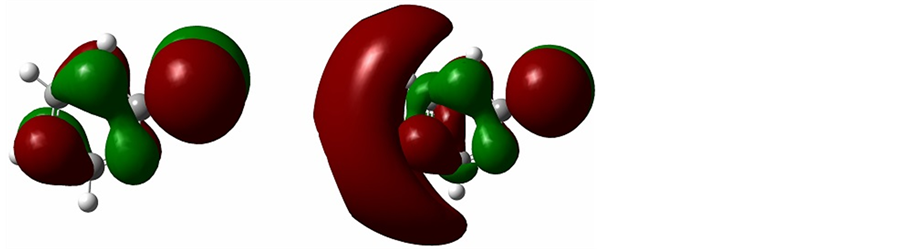
Figure 2. The 3D representation of HOMO and LUMO frontal orbitals of free (Dpds)-ligand
图2. 配体2-巯基吡啶离子前线轨道HOMO和LUMO的三维结构图
Figure 3. HOMO orbitals of complex
图3. 配合物前线轨道HOMO三维结构图
4. 结论
本文选用2,2’-二硫二吡啶(简称Dpds)这样一种含氮杂环化合物作为配体,以分层扩散法合成出六核铜簇合物。然后通过XRD单晶衍射对配合物进行结构测定。另外对配合物进行了量子化学计算和研究。本文的具体研究成果有:以Dpds为配体,采用分层扩散法,与铜离子反应,得到簇合物Cu6(C5H4NS)6。配合物属于单斜晶系,P2(1)/n空间群,每个分子含六个铜离子、六个2-巯基吡啶阴离子。配合物整个分子为1个分立的六核簇合物,六个2-巯基吡啶通过Cu-N、Cu-S配位键与Cu6核心结合。对配合物进行了量子化学计算研究,其计算结果与实验数据相符。
致谢
本论文得以完成,感谢唐豫媛对实验的指导,感谢国家自然科学基金及大学生创新项目的支持。
基金项目
国家自然科学基金(No. 21401151),四川省科技支撑计划(2015GZ0233),大学生国家创新项目(NO.201610613072)及省级创新项目(NO.2016099)。
文章引用
张连生,万 玮,王萃娟,熊霄阳,赵 鑫,李泽贤,陈志斌. 2-巯基吡啶配合物的合成及量子化学计算
The Synthesis and Quantum Chemistry Calculation Based on 2-Thiol Pyridine Ligand[J]. 有机化学研究, 2017, 05(02): 100-103. http://dx.doi.org/10.12677/JOCR.2017.52013
参考文献 (References)
- 1. Wang, X.F., Song, Q.S., Pan, Y., et al. (2016) Two New Cadmium Metal-Organic Frameworks Based on a Mixed-Donor Ligand. Chemical Research in Chinese Universities, 32, 539-544. https://doi.org/10.1007/s40242-016-5489-8
- 2. Chen, M.,Wang, Z.W., Sanudo, E.C., et al. (2014) Two Self-Interpenetrating Magnetic Mn(II) Metal-Organic Frameworks Assembled from Rigid or Flexible Tripodal Multicarboxylate Ligands. Inorganic Chemistry Communications, 43, 121-125. https://doi.org/10.1016/j.inoche.2014.02.021
- 3. 张江华, 吕英, 贾红亮, 等. 吲哚二甲川菁的合成, 晶体结构, 光谱性质及生物应用[J]. 高等学校化学学报, 2015, 36(10), 1924-1932.
- 4. Gallego, A., Castillo, O., Gómez-García, C.J., et al. (2011) Electrical Conductivity and Luminescence in Coordination Polymers Based On Copper (I)-Halides and Sulfur-Pyrimidine Ligands. Inorganic Chemistry, 51, 718-727.
- 5. Han, L., Bu, X., Zhang, Q., et al. (2006) Solvothermal in Situ Ligand Synthesis through Disulfide Cleavage: 3D (3,4)-Connectedand and 2D Square-Grid-Type Coordination Polymers. Inorganic Chemistry, 45, 5736-5738. https://doi.org/10.1021/ic0608041
- 6. 陈振峰, 王贤文, 黄凌, 等. 新法合成CU6(C5H4NS)6簇合物及其性质[J]. 应用化学, 2005, 22(11): 1268-1270.
- 7. Kitagawa, S., Munakata, M., Shimono, H., et al. (1990) Synthesis and Crystal Structure of Hexanuclear Copper (I) Complexes of µ3-Pyridine-2-Thionate. Journal of the Chemical Society, Dalton Transactions, No. 7, 2105-2109. https://doi.org/10.1039/DT9900002105
- 8. 陈敬中. 现代晶体化学理论与方法[M]. 北京: 高等教育出版社, 2011.
- 9. 祁景玉. X-射线结构分析[M]. 上海: 同济大学出版社, 2003.
- 10. Sheldrick, G.M. (1997) SHELXS, Program for Crystal Structure Solution. University of Göttingen, Germany,.
- 11. Sheldrick, G.M. (1997) SHELXL-97 Program for the Refinement of Crystal Structures. University of Göttingen, Germany.
- 12. Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al. (2008) Gaussian 03, Revision C.02.
- 13. Becke, A.D. (1993) Density-Functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics, 98, 5648-5652. https://doi.org/10.1063/1.464913
- 14. Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Physical Review B, 37, 785. https://doi.org/10.1103/PhysRevB.37.785
- 15. Vrkic, A.K., Taverner, T., James, P.F. and O’Hair, R.A. (2004) Gas Phase Ion Chemistry of Charged Silver(I) Adenine Ions via Multistage Mass Spectrometry Experiments and DFT Calculations. Dalton Transactions, No. 2, 197-208. https://doi.org/10.1039/b311656e
- 16. Mulliken, R.S. (1955) Electronic Population Analysis on LCAO-MO Molecular Wave Functions. IV. Bonding and Antibonding in LCAO and Valence-Bond Theories. The Journal of Chemical Physics, 23, 2343-2346. https://doi.org/10.1063/1.1741877
- 17. 钟建莲, 陈建华, 李玉琼, 等. 硫铁矿晶体化学及前线轨道研究[J]. 广西大学学报, 2011, 36(3): 405-409.
- 18. Wang, C.J., Wang, Y.Y., Ma, H.R., et al. (2006) A Novel Ligand N,N’-Di(2-Pyridyl)-2,4-diamino-6-phenyl-1,3,5- triazine(dpdapt) and Its Complexs: [Cu(dpdapt)Cl2] and [Cu(dpdapt)(NO3)(H2O)]∙NO3∙H2O[J]. Polyhedron, 25, 195- 202. https://doi.org/10.1016/j.poly.2005.06.061
- 19. Zhou, L.J., Luan, X.J., Wang, Y.Y., et al. (2006) Supramolecular Complexes Constructed with Carboxylate Cu(II) and 2-(2-Pyridyl)-Benzimidazole via Hydrogen Bonding. Journal of Coordination Chemistry, 59, 1107-112.