设为首页
加入收藏
期刊导航
网站地图
首页
期刊
数学与物理
地球与环境
信息通讯
经济与管理
生命科学
工程技术
医药卫生
人文社科
化学与材料
会议
合作
新闻
我们
招聘
千人智库
我要投搞
办刊
期刊菜单
●领域
●编委
●投稿须知
●最新文章
●检索
●投稿
文章导航
●Abstract
●Full-Text PDF
●Full-Text HTML
●Full-Text ePUB
●Linked References
●How to Cite this Article
Applied Physics
应用物理
, 2012, 2, 116-120
http://dx.doi.org/10.12677/app.2012.24020
Published Online October 2012 (http:/
/www.hanspub.org/journal/app.html)
Study of Spectroscopic Constants of Uranium Compounds and
Uranium Isotope Selectivity
Yunguang Zhang
School of Science, Xi’an University of Posts and Telecommunications, Xi’an
Email: zhangyunguang2008@yahoo.cn
Received: Jul. 5
th
, 2012; revised: Jul. 19
th
, 2012; accepted: Jul. 27
th
, 2012
Abstract:
Firstly, we calculated the spectroscopic constant of UF
6
molecule by using BLYP method and TZP basis set,
which are in well agreement with the experimental data. Secondly, we studied the photochemical reaction of UF
6
+ HCl
and the selectivity of uranium isotope under CO laser radiation and under CO
2
and CO laser radiation. The results show
there are better uranium isotope selectivity and response rate for CO
2
and CO laser radiation than for CO laser radiation.
Finally, we calculated molecular spectroscopic constant and isotope shift of U
2
F
6
, and we predict that the U
2
F
6
molecule
is better material of laser isotope separation than UF
6
.
Keywords:
Spectroscopic Constant; Isotope Shift; CO
2
Laser; CO Laser
铀化合物的光谱常数和铀同位素选择因子研究
张云光
西安邮电大学理学院,西安
Email: zhangyunguang2008@yahoo.cn
收稿日期:
2012
年
7
月
5
日;修回日期:
2012
年
7
月
19
日;录用日期:
2012
年
7
月
27
日
摘
要:
我们用
BLYP
密度泛函和
TZP
基组计算了
UF
6
分子的平衡结构和光谱常数,结果与实验值符合的很好;
接着我们计算了由
CO
激光单独辐射下以及由
CO
2
激光和
CO
激光共同辐射的
UF
6
+ HCl
光化学反应和铀同位
素的选择性,发现由
CO
2
和
CO
激光共同辐射
UF
6
+ HCl
反应时,能够使反应速率加快,会有更好的铀同位素
选择性;另外我们还计算研究了
U
2
F
6
分子的光谱常数和同位素位移,预测出
U
2
F
6
分子比
UF
6
分子更加适合做
激光分离铀同位素的原料。
关键词:
光谱常数;同位素位移;
CO
2
激光;
CO
激光
1.
引言
激光法分离铀同位素是激光技术和核技术结合
而产生的一种分离同位素的方法。由于该方法的分离
系数很高,经一次分离即可将天然铀浓缩到核电站堆
用浓缩铀的丰度,故具有投资小、耗点少、成本低和
生产规模灵活等优点
[1]
。激光分离铀同位素的理论研
究工作大致包括三个方面:分子结构和光谱的研究;
激光化学,分离机制的研究
[2]
。
为了获得良好的分离条件,应首先选择相应的具
有合适同位素位移光谱的分子,并对它的同位素分子
的光谱等性质做系统的研究。
UF
6
气体分子在铀同位
素分离中占据重要的地位,它在
50~100 K
左右,大
多数分子处于振动基态,同位素位移容易分辨
[3]
。在
过去几十年,人们在实验上和理论上对其振动光谱等
性质做了大量的研究。早在
1974
年,
Mcdowell
等人
就在实验上测量分析了
UF
6
分子的振动光谱数据
[4]
。
很多人还在实验上通过各种方法获得了
UF
6
分子的键
能
(UF
5
-F)
[5,6]
。但由 于
UF
6
的稀少性和放射性,在实验
Copyright © 2012 Hanspub
116
铀化合物的光谱常数和铀同位素选择因子研究
上研究它还是比较困难的,因此在理论上用量子化学
程序详细计算其物理化学性质就显的尤为重要。在本
文中,我们分别使用
BLYP
泛函和
TZP
基组计算了
UF
6
分子的平衡键长和振动频率。
自激光光化学法分离铀同位素问世以来,人们对
UF
6
与各种化合物的反应及激光对其影响的研究也日
趋增多
[7]
。很多科研工作者都在实验上研究了
UF
6
+
HCl
体系的热反应和激光反应动力学过程,结果显示
CO
和
CO
2
激光作用下的反应速率比热反应速率增加
了数倍
[8]
。在
20
世纪
70
年代,
Eerkens
第一次成功的
用一个可调谐的连续的
CO
2
激光器照射
UF
6
和
HCl
气体混合物中
235
UF
6
分子的吸收带
(
346
)
,使 其
处于激发态并与
HCl
发生反应生成固体
UF
4
,从而实
现铀同位素分离
[7]
。他们还计算出了其有效的铀浓缩
因子是
1.1
,这跟可能的最大的浓缩因子
1.5
符合的很
好
[9]
。
Eerkens
还在
1989
年用
CO
激光器照射
UF
6
/HCl
气体混合物中的
235
UF
6
分子的
3
振转带而获得浓缩
因子为
1.1
[10]
。本文中,我们首先在理论上研究了
CO
激光辐射下的
UF
6
+ HCl
光化学反应和铀同位素的选
择性,得到了
235
UF
6
分子的吸收带
3
3
的铀浓缩因子。
接着又研究了由
CO
2
和
CO
激光共同辐射
UF
6
+ HCl
反应的动力学过程,并计算得到了这个反应过程中的
分子吸收带
(4
346
)
的铀同位素浓缩因子。
另外值得注意的一个问题是同位素分子光谱中
的吸收峰的宽度常常受到诸如多普勒效应、压力加宽
以及分子间相互作用等影响而可能变的很宽,甚至会
完全掩盖掉原来就很小的同位素位移,使两个同位素
吸收峰彼此重叠,从而使分离难以实现。目前所广泛
使用的铀化合物
UF
6
分子的同位素位移仅为
0.6 cm
–1
左右,因此我们需要找到一种同位素位移更大的铀化
合物,以便更容易的进行铀同位素的分离。我们猜测
包含
U
2
的化合物会比包含
U
的化合物
UF
6
分子的同
位素位移更大。对于包含
U
2
的无机化合物没有任何
实验研究。在理论上
Gagliardi
和
Roos
通过多组态量
化方法研究了
U
2
分子中两个
U
原子的成键性质
[11]
。
他们发现
U
2
分子有比其它所有可知的双原子分子的
化学键都复杂的五重键。根据这个性质,在理论上可
以形成无机
U
2
F
6
分子。本文中,我们用全电子密度泛
函方法计算了
U
2
F
6
分子的几何结构、振动频率和同位
素位移,在理论上预测
U
2
F
6
可能比
UF
6
更加适合做铀
同位素分离的原料。
2.
计算方法
本文中,计算采用的是密度泛函方法,选用了
BLYP
泛函。密度泛函理论是一种研究多电子体系电
子结构的量子力学方法,在物理方面有广泛的应用。
电子结构理论的经典方法,特别是
Hartree-Fock
,是
基于复杂的多电子波函数的。密度泛函理论的主要目
标就是用电子密度取代波函数作为研究的基本量。因
为多电子波函数有
3N
个变量
(N
为电子数,每个电子
包含三个空间变量
)
,而电子密度仅是三个变量的函
数,所以更加方便处理。密度泛函理论最普遍的应用
是通过
Kohn-Sham
方法实现
[12]
。在这个方法中,最难
处理的多体问题被简化成了一个没有相互作用的电
子在有效势场中运动的问题。这个有效势场中包括了
外部势场以及电子间库仑相互作用的影响,例如交换
和相关作用。自
1970
年以来,密度泛函理论在量化
计算中得到广泛的应用。在多数情况下,与其它解决
量子力学多体问题的方法相比,采用密度泛函理论给
出了非常令人满意的结果,且要比用其它方法更节省
时间。尽管如此,人们普遍认为它不能给出足够精确
的结果,直到二十世纪九十年代,理论中所采用的近
似被重新提炼成更好的交换相关作用模型。密度泛函
理论是目前多种领域中电子结构计算的领先方法。本
章中计算所用的程序是
ADF2009
软件包
[13]
。
3.
结果与分析
3.1. UF
6
分子结构和振动频率
在本文中,我们使用
BLYP
泛函和
TZP
基组计算
了
UF
6
分子的平衡键长和振动频率。其中标量和相对
论效应通过
ZORA
方法引入狄拉克方程
[14]
。此外我们
在保证计算精确度的情况下对
F
和
U
原子采取冰冻核
电子的方法,分别为:
F.1s
和
U.5d
。
计算结果表明
UF
6
分子属于单重态。平衡键长
(U-F)
计算值为
1.9986 Å
,与实验值
1.996 Å
[15]
符合的
很好,误差仅为
0.13%
。八面体结构的
UF
6
分子有六
个振动模型:
T
mol
= A
1g
+ E
g
+ 2T
1u
+ T
2g
+ T
2u
。在这四
种模型中只有
T
1u
(
3
和
4
)
是红外活性,其中
3
和
4
分别对应反对称伸缩和弯曲模型。其计算结果列于表
1
中。
从这个表中我们可看到,用
BLYP
方法计算的振
动频率的结果与实验值符合的很好。例如,
3
振动频
率计算值为
628.4782 cm
–1
,实验值为
626 cm
–1
,它 们
Cop
yright © 2012 Hanspub
117
铀化合物的光谱常数和铀同位素选择因子研究
Table 1. The calculation result of vibration frequency of UF
6
表
1. UF
6
分子的振动频率计算结果
Method
1
2
3
4
5
6
BLYP 669.2797 533.8869 623.6415181.4717 191.4623 139.7444
Expt
[4]
667 534 626 186 200 143
的差距仅为
2.4782 cm
–1
。另 外
UF
6
分子的六个振动模
型可以被分为三个伸展模型
(
1
,
2
和
3
)
和三个弯曲
模型
(
4
,
5
和
6
)
。
3.2. CO
和
CO
2
激光催化光化反应的铀同位素
分离
由
CO
和
CO
2
激光共同作用
235
UF
6
分子的吸收带
(
346
)
和
3
带,使其到达激发态与
HCl
发生反
应生成固体
UF
4
,从而分离出
235
U
。为了计算上述反
应下的浓缩因子,我们首先需要分析由
CO
单独作用
235
UF
6
分子吸收带
(
346
)
的情况。
从此分析中能
够得到一些必要的信息。
对于
CO
激光分离铀同位素的情况,我们给出一
组速率方程:
11
AaAA
Ng kNgN
12
A
22
A
2
(3.1)
22
AbAA
Ng kNgN
(3.2)
13
1132
3
3
A
aAAvvAvMBLB a
N
kNgNkNkN kN k
(3.3)
23
2232
3
3
A
bAAvvAvMB LB b
N
kN gNkNkN kN k
(3.4)
,
,0, ,0,1,
AiLi B AiLi Ai
LiLia bLiTa b
NkNNKN
KKkk KKkki
(3.5)
其中, ,和
1
A
N
2
A
N
B
N
分别是
235
UF
6
,
238
UF
6
和
HCl
分子的密度;
1
L
K
和
2
L
K
是
CO
激光作用下的反应速
率;
T
K
是热反应速率; 和 是选择性激发速率;
, 和
a
k
b
k
vv
k
vM
k
L
k
分别是振动–振动转移,碰撞转移和
光敏反应速率系数。另外自发辐射被忽略。所有的计
Table 2. Isotopic selectivity
reaction of UF
算结果列
于表
2
中。
of
CO laser-catalyzed photochemical
表
Intracavity power100 W 200 W 300 W
6
+ HCl (T = 253 K)
2. CO
激光催化光化学反应
UF
6
+ HCl (T = 253 K)
的铀同位素选
择因子
I
co
1
−
2
20
−
2
3
00 W·cm
0 W·cm
00 cm
−
2
235
σ
238
σ
2 × 10
cm
(1/1.5)
235
σ
−
22
2
2
2
t t = 1 s)
2 × 10
cm
(1/1.5)
235
σ
−
22
2 × 10
cm
(1/1.5)
235
σ
−
22
0.036 s
−
1
K
L
0.036 s
−
1
0.016 s
−
1
0.036
−
1
s
0.016 s
−
1
0.016 s
−
1
K
T
K
0.036 s
−
1
0.036 s
−
1
V-V
K
0.036 s
−
1
0.0036
−
1
V-M
β
(1)(a
0.0036
-1
s
1.08149
0.0036
−
1
s
1.04158
s
1.02797
β
(10)(at t = 10 s)
β
(20)(at t = 20 s
1.07240 1.0376 1.02546
)1.0655 1.03459 1.02351
双激光系统
(CO
和
CO
2
)
的光化学反应模型主要
的激
6
光激
发过程如下:
1
k
2
235
235 *
6co 634
UF UF
a
gh
(3.6)
2
235*235 *
6346CO6 346
UFUF 4
a
k
h
(3.7)
1
235
235 *
6co 6
UFUF 3
a
k
gh
3
(3.8)
2
2
235
235 **
63 co6346
UF 3UF 4
a
k
hv
(3.9)
6
3
2
238
238 *
6co 634
UF UF
a
k
gh
(3.10)
4
238
238 **
6346co6 346
UFUF 4
a
k
h
(3.11)
3
238
238 *
66
UFUF 3
a
k
co
gh
3
(3.12)
4
2
238
238 **
63 co6346
UF 3UF4
a
k
h
(3.13)
其中
, ,
1
a
k
2
a
k
1
a
k
,
2
a
k
, , , 和
3
a
k
4
a
k
3
a
k
4
a
k
分别
过的 择有 计
CO
2
和
CO
激光比单独使用
CO
激
光催
是各自对
应 程中 选 性激发几率。所 的 算结
果列于表
3
中。
可见共同使用
化
UF
6
+ HCl
反应得到的铀同位素浓缩因子高。
当激光强度
2
CO
200 Wcm
I
和温度
T = 253 K
时,
铀同位素选择
(1) 1.04158
性因子分别为
和
(10)
1.0376
;当激光强度
300 Wcm
I
,
2
和相同温
别为
(1) 1
CO
选择性因子分
度时
.02797
铀同
位素
和
(10)1.02546
。当
2
2
cm
II
时
(T = 235
K)
,共同使用
CO
2
和
CO
激光催化
UF
可
(1
CO
CO
性因子
) 4.05338
50 W
6
+ HCl
反应
得到选择
。激光强度是影响铀同
位素选择性的重要因素,其中强度为
50~100 W·cm
−
2
Cop
yright © 2012 Hanspub
118
铀化合物的光谱常数和铀同位素选择因子研究
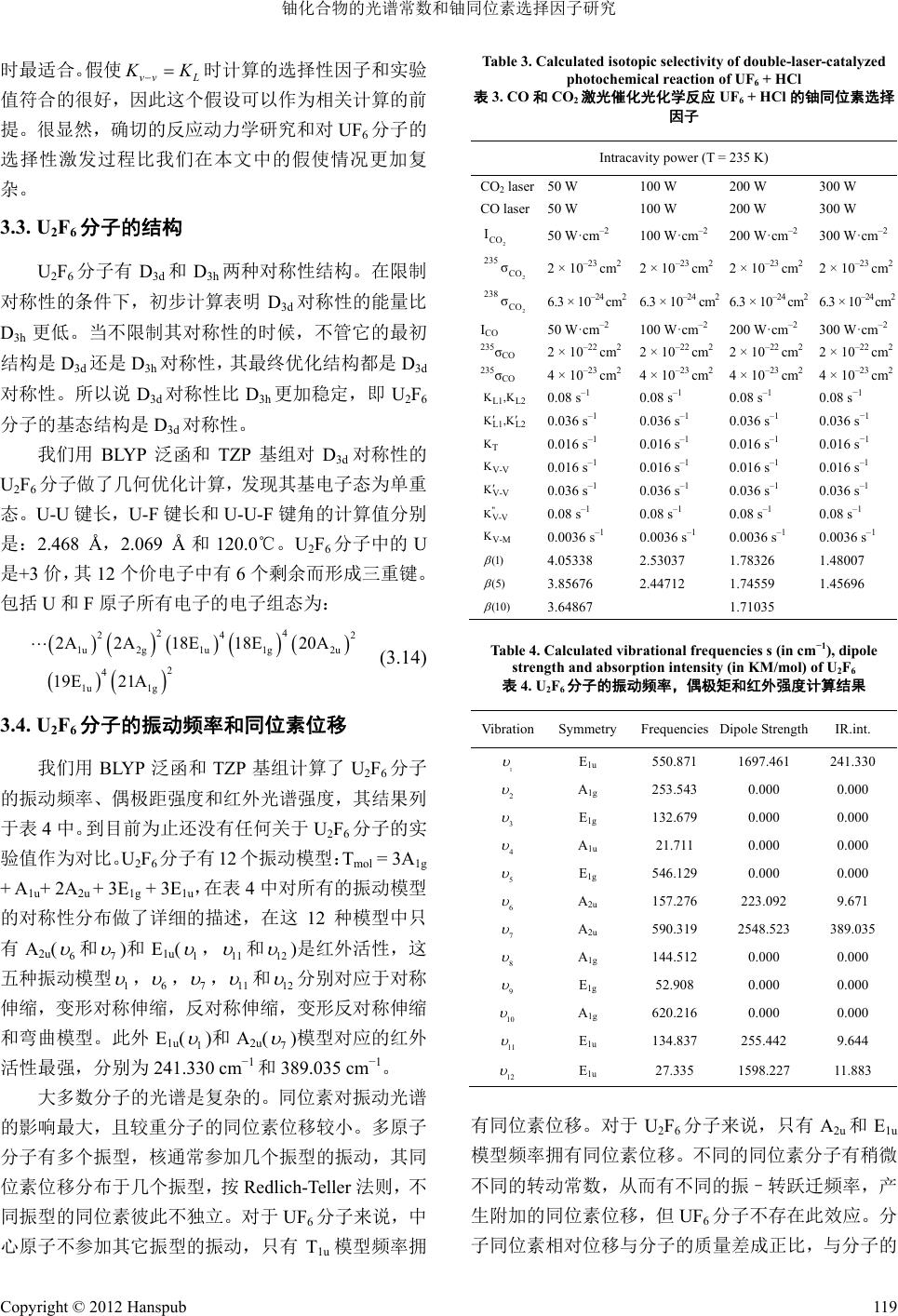
时最适合。
假使
vv L
K
K
的反应
D
和
D
下,初步计算
时计
动
分子有
3h
两种对称性结构。在限制
表明
D
3d
对称性的能量比
D
3h
其基电子态为单重
态。
算的选择性因子和实验
值符合的
很好,因此这个假设可以作为相关计算的前
提。很显然,确切力学研究和对
UF
6
分子的
选择性激发过程比我们在本文中的假使情况更加复
杂。
3.3. U
2
F
6
分子的结构
U
2
F
6 3d
对称性的
条件
更低。当不限制其对称性的时候,不管它的最初
结构是
D
3d
还是
D
3h
对称性,
其最终优化结构都是
D
3d
对称性。所以说
D
3d
对称性比
D
3h
更加稳定,即
U
2
F
6
分子的基态结构是
D
3d
对称性。
我们用
BLYP
泛函和
TZP
基组对
D
3d
对称性的
U
2
F
6
分子做了几何优化计算,发现
U-U
键长,
U-F
键长和
U-U-F
键角的计算值分别
是:
2.468 Å
,
2.069 Å
和
120.0
℃。
U
2
F
6
分子中的
U
是
+3
价,其
12
个价电子中有
6
个剩余而形成三重键。
包括
U
和
F
原子所有电子的电子组态为:
24
1g
21A
YP
泛函和
24
(3.14)
3.4. U
2
F
6
分子的振动频率和同位素位移
BL
TZP
基组计算了
U
2
F
6
分子
其结果列
于表
2
1u 2g1u1g2u
2
4
2A2A18E 18E20A
1u
19E
我们用
的振动频率、偶极距强度和红外光谱强度,
4
中。
到目前为止还没有任何关于
U
2
F
6
分子的实
验值作为对比。
U
2
F
6
分子有
12
个振动模型:
T
mol
= 3A
1g
+ A
1u
+ 2A
2u
+ 3E
1g
+ 3E
1u
,
在表
4
中对所有的振动模型
的对称性分布做了详细的描述,在这
12
种模型中只
有
A
2u
(
6
和
7
)
和
E
1u
(
1
,
11
和
12
)
是红外活性,这
五种振动模型
1
,
6
,
7
,
11
和
12
分别对应于对称
伸缩,变形对 伸缩,对变形反对称伸缩
和弯曲模型。此外
(
1
称 反
E
1u
称伸缩,
)
和
A (
7
2u
)
模型对应的红外
活性最强,分别为
241.330 cm
−
1
和
389.035 cm
−
1
。
大多数分子的光谱是复杂的。同 素对振动光谱
的影响最大,且较重分子的同位素位移较小。多原子
位
分子有多个振型,核通常参加几个振型的振动,其同
位素位移分布于几个振型,按
Redlich-Teller
法则,不
同振型的同位素彼此不独立。对于
UF
6
分子来说,中
心原子不参加其它振型的振动,只有
T
1u
模型频率拥
Table 3. Calculated isotopic sele
ctivity of double-laser-catalyzed
photochemical reaction of UF
6
+ HCl
表
3. CO
和
CO
2
激光催化光化学反应
UF
6
+ HCl
的铀同位素选择
因子
Intracavity power (T = 235 K)
CO
2
laser50 W 100 W 200 W 300 W
CO laser50 W 300 W
m
10m
20m
–2
30m
–2
3
cm
2
3
cm
2
3
cm
2
3
cm
2
2 2
2
2
I
CO
235
σ
CO
235
σ
100 W 200 W
–2 –2
2
CO
I
50 W·c
0 W·c
0 W·c
0 W·c
2
235
CO
σ
2 × 10
–2
2 × 10
–2
2 × 10
–2
2 × 10
–2
2
238
CO
σ
6.3 × 10
–24
cm
6.3 × 10
–24
cm
6.3 × 10
–24
cm
6.3 × 10
–24
cm
50 W·cm
–2
100 W·cm
–2
200 W·cm
–2
300 W·cm
–2
2 × 10
–22
cm
2
–23
2
2 × 10
–22
cm
2
–23
2
2 × 10
–22
cm
2
–23
2
2 × 10
–22
cm
2
–23
2
CO
K,
4 × 10
cm
0.08 s
–1
4 × 10
cm
0.08 s
–1
4 × 10
cm
0.08 s
–1
4 × 10
cm
0.08 s
−
1
L1 L2
,K
K
L1 L2
K
0.036 s
0.
0.
–1
036 s
–1
0.036 s
0.
0.
–1
036 s
1
−
1
T
K
0.016 s
–1
–1 –1
0.
–1
−
016 s
–1
016 s
–1
016 s
–1
V-V
K
0.016 s
0.016 s
0.016 s
0.016 s
V-
K
V
0.036 s
0.
–1
–1
036 s
0.
–1
–1
036 s
0.
–1
–1
036 s
–1
–1
"
V-V
K
0.08 s
–1
0.08 s
–1
0.08 s
–1
0.08 s
–1
V-M
K
0.0036 s
0.
0. 0.
0036 s
0036 s0036 s
(1)
4.05338 2.53037 1.78326 1.48007
(5)
3.85676 2.44712 1.74559 1.45696
(10)
3.64867 1.71035
Tad vibrational frequme
renbsorpon intensity ol) o
2
F
6
表
4. U
2
F
6
分子的振动频率,偶极矩和红外强度计算结果
Vibra
ble 4. Calculateencies s (in c
−
1
), dipol
stgth and ati(in KM/mf U
tionSymmetryFrequencies Dipole Strength IR.int.
1
E
1u
550.871 1697.461 241.330
2
A
1g
253.543 0.000 0.000
3
E
1g
132.679 0.000 0.000
4
A
1u
21.711 0.000 0.000
5
E
1g
546.129 0.000 0.000
6
A
2u
157.276 2 23.0929.671
7
A
2u
590.319 2 3548.52389.035
8
A
1g
144.512 0.000 0.000
9
E
1g
52.908 0.000 0.000
10
A
1g
620.216 0.000 0.000
11
E
1u
134.837 2 55.4429.644
12
E
1u
27.335 1 598.227 11.883
有同位素位移于
U
子来
A
1u
模型频率拥有同位素位移。不同的同位素分子有稍微
。对
2
F
6
分说,只有
2u
和
E
不同的转
动常数,从而有不同的振–转跃迁频率,产
生附加的同位素位移,但
UF
6
分子不存在此效应。分
子同位素相对位移与分子的质量差成正比,与分子的
Cop
yright © 2012 Hanspub
119
铀化合物的光谱常数和铀同位素选择因子研究
Copyright © 2012 Hanspub
120
UF
6
和
U
2
F
6
分子的各自最大的同位素位移。对于
质量成反比。分子的质量越大,其相对位移量越小。
同时还说明质量大的同位素分子比质量小的同位素
分子频率
小。在自然界中
U
原子主要有两个稳定的同
位素
238
U
和
235
U
。在实验上人们也有测量出了
238
UF
6
和
235
UF
6
同位素分子的位移。在本文中,我们重点分析
了
UF
6
分子,反对称伸缩频率
3
的同位素位移是
Δ
(
235
UF
6
–
238
UF
6
) = 0.714 cm
−
1
。
U
2
F
6
分子的对称伸
缩频率
7
的同位素位移为
Δ
(
235
U
2
F
6
–
238
U
2
1.
739 cm
−
1
。很明显
U
2
F
6
分子的同位素位移要比
UF
6
分子大的 ,而同位素位移的大小对激光分离同位素
有至关重要的作用。所以我们根据计算结果预测出
U
2
F
6
分子有可能是比
UF
6
分子更加适合的激光分离同
位素的原料。当然这还需要人们对其进行更深入的研
究。
4.
结论
F
) =
多
用
BLYP
泛函和
TZP
基组计算了
UF
2 6
衡键长和振动频率。通过与已有的实
值对
6
我们使
U F
分子的平
6
和
验
比可
知计算得到的光谱数据误差较小,可见用此
方法研究铀化合物的性质是可行的。另外我们在理论
上研究了
CO
激光辐射下的
UF
6
+ HCl
光化学反应和
铀同位素的选择性,得到了
235
UF
6
分子的吸收带
3
3
的铀浓缩因子。接着又研究了由
CO
2
和
CO
激光共同
辐射
UF
6
+ HCl
反应的动力学过程,并计算得到了这
个反应过程中的分子吸收带
(4
346
)
的铀同位
素浓缩因子。结果表明由
CO
2
和
CO
激光共同辐射
UF
6
+ HCl
反应时能够使反应速 会有更好的
铀同位素选择性,即同时用
CO
和
CO
2
激光要比单独
用其中的一种激光获得的铀浓缩因子高,其原因需要
进一步研究。最后我们用相同方法对
U
2
F
6
分子的振动
频率和同位素位移做了计算,发现
U
2
F
6
分子的
12
种
模型中只有
A
2u
(
6
率加快,
和
7
)
和
E
1u
(
1
,
11
和
12
)
是红外
活性,其中
1
和
7
振动模型对 的红外活性最强。此
外
U
6
分的同位素位移计算值要比
UF
6
分子大的
多,而同位素位移的大小对激光分离同位素有至关重
要的作用。所以我们预测
U
2
F
6
分子有可能是比
UF
6
分子更加适合的激光分离同位素的原料。
应
原子能出
2
F
参考文献
)
7(5):
子
(References
[1]
肖啸菴
.
同位素分离
[M].
北京
:
版社
, 1999.
[2]
吕百达
,
匡一中
.
激光分离同位素
[J].
激光杂志
, 1986,
284-290.
[3]
宋文忠
,
古端
.
六氟化铀低温红外光谱
[J].
核
化学与放射化
学
, 1990, 12(3): 175-179.
[4]
R. S. McDowell, L. B. Asprey and R. T. Paine. Vibrational spectrum
and force field of uranium hexafluoride. Journal of Chemical
Physics, 1974, 61(1): 3571-3580.
[5]
D. L. Hildenbrand, K. H. Lau. Redetermination of the thermo-
chemistry of gaseous UF
, UF
, a
52
nd UF. Journal of Chemical
Physics, 1991, 94(2): 1420-1425.
[6]
R. N. Compton. On the formation of positive and negative ions
in gaseous UF
.
Jo
urnal of Chemica
6
l Physics, 1977, 66(10):
4478-
4485.
[7]
J. M. Eerkens. D
6
Reaction chemistry of the UF
6
lisosep process.
Optics Communications, 1976, 18(1): 32-33.
[8]
徐葆裕
,
胡建勋
,
郑成法
.
六氟化铀与卤化氢气体的反应动
力学研究
[J].
化学学报
, 1997, 55(10): 979-982.
[9]
J. W. Eerkens. Spectral considerations in the laser isotope separation
of Uranium Hexafluoride. Applied Physics A, 1976, 10(1): 15-
31.
[10]
J. W. Eerkens. International Uranium Enrichment Conference.
California: Monterrey, 1989.
[11]
L. Gagliardi, B. O. Roos. Quan
tum chemical calculations show
that the uranium molecule U
h
2
as a quintuple bond. Nature,
2005,
433: 848-851.
[12]
G. Te, Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J.
A. van Gisbergen, J. G. Snijders and T. Ziegler.
Chemistry with
ADF
.
Journal of Computational Chemistry, 2001, 22(9): 931-
967.
[13]
J. P. Perdew, K. Burke and K. M. Ernzerhof. Generalized gradient
approximation made simple. Physical Review Letters, 1996, 77
(18): 3865-3868.
[14]
E. van Lenthe, E. J. Baerends and J. G. Snijders. Relativistic
regular two component Hamiltonians. Journal of Chemical Physics,
1993, 99(6): 4597-4610.
[15]
M. Kimura, V. Schomaker, D. M. Smith and B. Weinstock. Electron
diffraction investigation of the hexafluorides of tungsten, osmium,
iridium, uranium, neptunium, and plutonium. Journal of Chemical
Physics, 1967, 48(9): 4001-4012.