 Hans Journal of Biomedicine 生物医学, 2013, 3, 1-6 http://dx.doi.org/10.12677/hjbm.2013.31001 Published Online January 2013 (http://www.hanspub.org/journal/hjbm.html) Conformation Transformation of Aβ42 Protein under Different Temperature by Molecular Dynamics Simulations* Jing Fu1,2, Liling Zhao1,3, Jihua Wang1,3# 1Shandong Provincial Key Laboratory of Functional Macromolecular Biophysics, Dezhou University, Dezhou 2College of Physics and Electronics, Shandong Normal University, Jinan 3Department of Physics, Dezhou University, Dezhou Email: fujing0203@126.com, zhaoll@sina.com, #jihuawang169@gmail.com Received: Nov. 21st, 2012; revised: Dec. 18th, 2012; accepted: Dec. 27th, 2012 Abstract: The temperature-induced conformation changes of Aβ42 protein were studied by molecular dynamics simu- lation. The three independent molecular dynamics simulations of Aβ42 protein at different temperatures 300 K, 340 K and 380 K, were performed using the GROMACS software package and GROMOS 43A1 force field, respectively. Each simulation was run for 60 ns. Based on the simulations, we analyzed the conformation changes of Aβ42 protein and the formation of its secondary structure and tertiary structure. The results indicated that Aβ42 protein has no stable structure and it has the characters of intrinsically disorder proteins at the different temperatures. It also shows that the structure of Aβ42 protein change obviously with different temperature. In addition, there is a change tendency from α-helix to β-sheet at 380 K. Keywords: Aβ42 Protein; Molecular Dynamics Simulation; Alzheimer’s Disease; Conformation Transformation 基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化* 付 静1,2,赵立岭 1,3,王吉华 1,3# 1德州学院山东省功能大分子生物物理重点实验室,德州 2山东师范大学物理与电子科学学院,济南 3德州学院物理系,德州 Email: fujing0203@126.com, zhaoll@sina.com, #jihuawang169@gmail.com 收稿日期:2012 年11 月21 日;修回日期:2012年12 月18 日;录用日期:2012年12 月27 日 摘 要:基于分子动力学模拟方法研究了不同温度对 Aβ42 蛋白结构的影响。模拟选用 GROMACS 软件包和 GROMOS 43A1分子力场,在 300 K、340 K 和380 K 下分别进行 60 ns的分子动力学模拟。通过计算原子均方 根涨落、回旋半径、二级结构形成几率等参数,分析了Aβ42 蛋白结构的稳定性、二级结构及三级结构形成。 研究发现:该蛋白在三种不同温度下,都没有稳定结构,表现出固有无序特征,温度会导致结构特征发生显著 变化;在高温(380 K)时,会产生 α螺旋向 β折叠转换的趋势。 关键词:Aβ42 蛋白;分子动力学模拟;阿尔兹海默症;构象转变 1. 引言 阿尔兹海默症(Alzheimer’s Disease, AD)是一种 渐进性大脑退行性疾病,是当今威胁老年人身心健 康的三大疾病(癌症、心血管疾病和AD)之一。其发 病率是随年龄增长而显著升高的,65~80 岁人群约为 1%~5%,80岁以上者可达20%~40%[1]。因此,对于 AD 发病机理和防治的研究近年来成为国内外关注 的热点。 *基金项目:国家自然科学基金项目(30970561 和31000324)和山东 省自然科学基金项目(2009ZRA14028)。 #通讯作者。 Copyright © 2013 Hanspub 1  基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化 主流观点认为,AD病与淀粉样不溶性纤维的形 成密切相关[2],其微观结构是 β-淀粉样蛋白形成的反 平行 β片组成。β-淀粉样蛋白(Aβ)是一种含有 39~43 个氨基酸的多肽,是淀粉样蛋白前体蛋白跨膜分泌 APP 的产物[3]。其中,具有 40 和42 个氨基酸的肽段 被广泛研究,这是因为Aβ40 是脑质膜和脑脊液中的 正常可溶性产物,而Aβ42 由于其疏水性而认为在淀 粉样斑块的成核过程中发挥重要作用[4]。Aβ具有很强 的自聚性,一旦发生,很快形成极难溶解的沉淀,Aβ42 比Aβ40 聚集更快,为老年斑的主要成分。因此,通 过对 Aβ42 的研究对认识 AD 病的微观致病机制有很 重要的实际意义。 已有研究表明[2]Aβ在聚集状态下以β-折叠为主, 而非聚集时则根据不同的环境采用不同构象,如在生 物膜和有机溶剂中以 α-螺旋为主,在水溶液中则以卷 曲为主。由于Aβ非常容易聚集,且构象转变所需时 间非常短,因此到目前为止仍无法通过实验手段检测 Aβ从α-螺旋到β-折叠或者从卷曲到β折叠的构象转 变过程,而计算机模拟是研究其构象变化的重要工具 [5,6],Xu 等[7]用分子动学模拟方法研究了 Aβ40 从α- 螺旋结构到 β-折叠结构构象转换的机制,Wei 等[8]利 用副本交换分子动力学模拟分析了 Aβ(25-35) 肽段二 聚物的结构特征。 β-折叠结构可促进 Aβ聚集成不溶性纤维,不易 被蛋白酶降解,进一步形成极难溶解的不溶性纤维, 并由此生成老年斑。Aβ由可溶状态到 不溶 状态的 转 变是 AD 发病机理中的关键因素[9],该过程中涉及许 多因素,主要与分子的疏水性、电荷数等特点有关, 也受 pH 值、温度、金属离子等因素影响。Aβ的C端 最后几个氨基酸有很强的疏水性,且 C端越长越易聚 集成纤维状。研究发现[10],在Aβ潜伏期的早期有纤 维状部分存在。Aβ42 被认为是引起 AD病产生的重要 原因,主要是因为它的错误折叠和聚集,小的低聚物 能导致很强的毒性。因此,阻止Aβ42的错误折叠/聚 集,或者两者之一从而减少Aβ42 低聚物的形成,对 AD 病的治疗有重要意义[11]。 尽管实验生物学对Aβ42 作了大量研究和统计, 对Aβ42 蛋白聚集引起的结构变化等进行了研究。但 是在原子层次,不同温度对 Aβ42 构象变化有何影响, 温度是否影响Aβ聚集等问题尚不清楚。分子动力学 模拟为研究此类问题提供了强有力的工具,它已经成 为生物大分子结构功能关系研究方面不可缺少的重 要工具,在蛋白质折叠、去折叠及其与疾病关系方面 开展了大量研究工作[5-7,12]。本文利用分子动力学模拟 方法,研究Aβ42 蛋白质在不同温度下构象变换情况。 研究结果表明,该蛋白在三种不同温度均没有稳定的 三级结构,温度对二级结构和β-sheet 的形成有明显影 响,这对Aβ42 微观聚集的温度效应认识有重要意义。 2. 材料和方法 Aβ42 蛋白的初始构象取自蛋白 质结构数据库 (PDB 号为 1IYT),如 图1所示[13]。该结构中含有两段 螺旋,分别为 helix-1(残基 7~26)和helix-2(残基27~ 40),是在膜环境下的结构。利用 GROMACS 软件包 [14]和GROMOS43A1 力场[15],分别对体系在恒温恒压 (NTP)系综下进行分子动力学模拟。溶剂选用显含水 的单点电荷(SPC)模型,蛋白放在周期性、大小合适 的十二面体中,溶质同盒子边缘最小间距设定为 0.8 nm。带电基团选用残基在 pH 值为 7.0 下的质子化状 态。采用反应场修正的双阈值法来处理原子之间的非 键相互作用,即对距离小于0.8 nm的短程相互作用, 每步进行计算,距离介于 0.8 nm和1.4 nm之间的中 程相互作用,每 10步更新一次,对距离超过 1.4 nm 的静电相互作用,利用连续介质反应场近似来处理, 其中水的相对介电常数为 54。用SHAKE 算法[16]固定 所有的共价键,相对偏差设为10–4。利用最陡下降法 进行 1000 步能量优化,以消除不合理的能量势垒。 为使模拟体系保持电中性,Aβ42 蛋白体系加入 3个 带正电的钠离子,然后进行第二次能量优化。从初始 构象出发,约束溶质原子,进行 300 ps 的约束动力学 模拟,通过逐步升温的方法分别将体系温度由 50 K 上升到 300 K、340 K和380 K。然后在各自温度下分 别进行 60 ns 的自由分子动力学模拟。 Figure 1. The structure of Aβ42 (PDB entry 1ITY) 图1. Aβ42 的结构(PDB 代号为 1IYT) Copyright © 2013 Hanspub 2 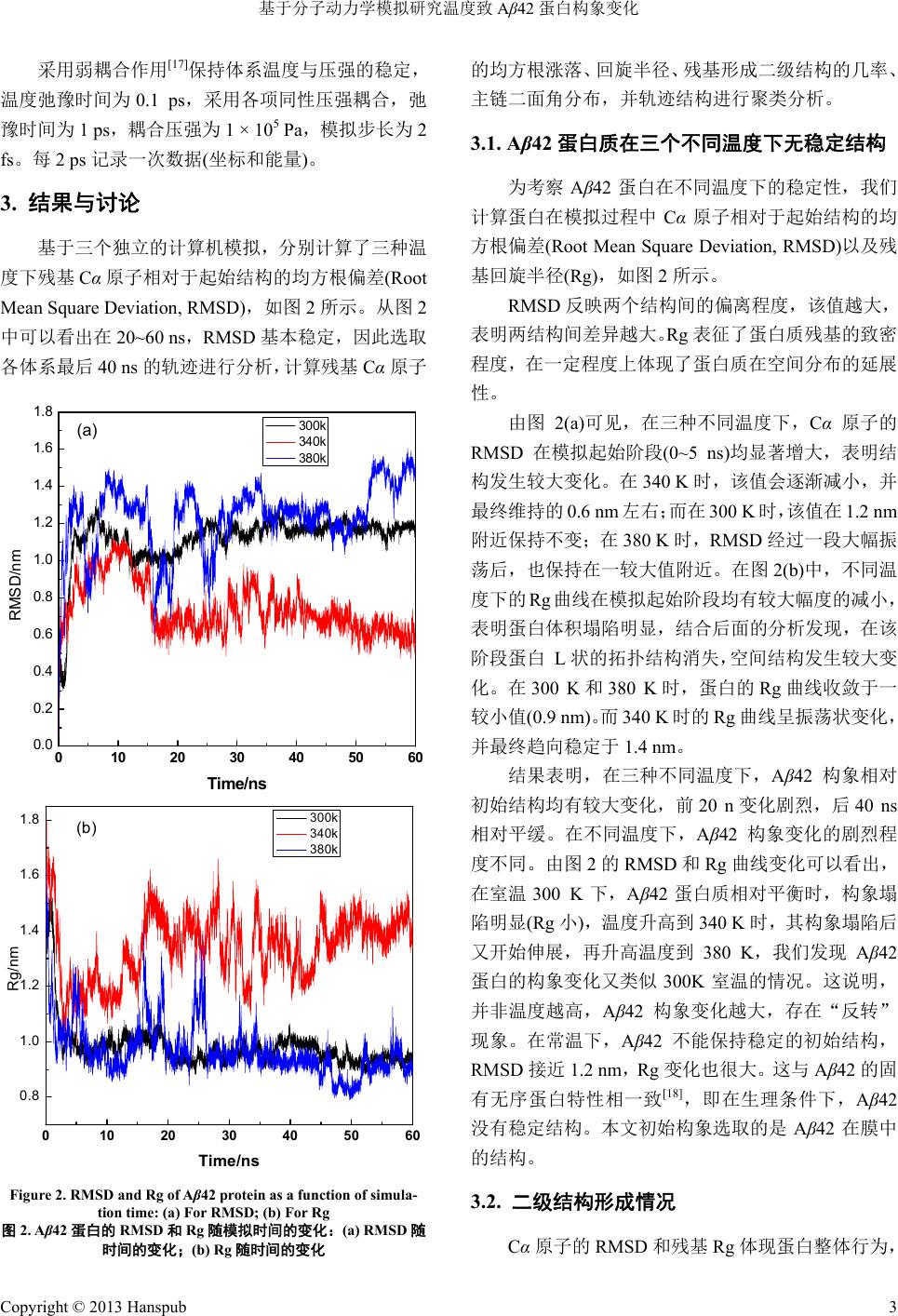 基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化 采用弱耦合作用[17]保持体系温度与压强的稳定, 温度弛豫时间为 0.1 ps,采用各项同性压强耦合,弛 豫时间为1 ps,耦合压强为1 × 105 Pa,模拟步长为 2 fs。每 2 ps 记录一次数据(坐标和能量)。 3. 结果与讨论 基于三个独立的计算机模拟,分别计算了三种温 度下残基 Cα原子相对于起始结构的均方根偏差(Root Mean Square Deviation, RMSD),如图 2所示。从图 2 中可以看出在20~60 ns,RMSD基本稳定,因此选取 各体系最后 40 ns 的轨迹进行分析,计算残基 Cα原子 0 10203040 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 5060 RMSD/nm Time/ns 300k 340k 380k (a) Rg/nm 0 0 10203040 0.8 1.0 1.2 1.4 1.6 1.8 5060 Rg/nm Time/ns 300k 340k 38 (b) 0k Figure 2. RMSD and Rg of Aβ42 protein as a function of simula- 图蛋白 随 的均方根涨落 构的几率、 3.1. Aβ42 蛋白质在三个不同温度下无稳定结构 为考察 Aβ42 蛋白在不同温度下的稳定性,我们 计算 该值越大, 表明 图2(a)可见,在三种不同温度下,Cα原子的 RMSD 度下,Aβ42 构象相对 初始 3.2. 二级结构形成情况 Cα原子的 RMSD 和残基 Rg 体现蛋白整体行为, tion time: (a) For RMSD; (b) For Rg 2. Aβ42 的RMSD 和Rg 随模拟时间的变化:(a) RMSD 时间的变化;(b) Rg 随时间的变化 、回旋半径、残基形成二级结 主链二面角分布,并轨迹结构进行聚类分析。 蛋白在模拟过程中 Cα原子相对于起始结构的均 方根偏差(Root Mean Square Deviation, RMSD)以及残 基回旋半径(Rg),如图 2所示。 RMSD 反映两个结构间的偏离程度, 两结构间差异越大。Rg 表征了蛋白质残基的致密 程度,在一定程度上体现了蛋白质在空间分布的延展 性。 由 在模拟起始阶段(0~5 ns)均显著增大,表明结 构发生较大变化。在 340 K 时,该值会逐渐减小,并 最终维持的 0.6 nm左右;而在300 K时,该值在 1.2 nm 附近保持不变;在 380 K 时,RMSD经过一段大幅振 荡后,也保持在一较大值附近。在图2(b)中,不同温 度下的 Rg 曲线在模拟起始阶段均有较大幅度的减小, 表明蛋白体积塌陷明显,结合后面的分析发现,在该 阶段蛋白 L状的拓扑结构消失,空间结构发生较大变 化。在 300 K和380 K时,蛋白的 Rg曲线收敛于一 较小值(0.9 nm)。而340 K 时的Rg 曲线呈振荡状变化, 并最终趋向稳定于 1.4 nm。 结果表明,在三种不同温 结构均有较大变化,前20 n变化剧烈,后 40 ns 相对平缓。在不同温度下,Aβ42 构象变化的剧烈程 度不同。由图 2的RMSD和Rg 曲线变化可以看出, 在室温 300 K下,Aβ42 蛋白质相对平衡时,构象塌 陷明显(Rg 小),温度升高到 340 K 时,其构象塌陷后 又开始伸展,再升高温度到380 K,我们发现 Aβ42 蛋白的构象变化又类似300K室温的情况。这说明, 并非温度越高,Aβ42 构象变化越大,存在“反转” 现象。在常温下,Aβ42 不能保持稳定的初始结构 , RMSD 接近1.2 nm,Rg 变化也很大。这与Aβ42 的固 有无序蛋白特性相一致[18],即在生理条件下,Aβ42 没有稳定结构。本文初始构象选取的是 Aβ42 在膜中 的结构。 Copyright © 2013 Hanspub 3  基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化 2013 Hanspub 要得 ln 到温度对蛋白局部结构影响,还需要对二级结构 情况进行分析。为此,我们利用 stride 程序[19]计算了 Aβ42 各残基形成二级结构的几率,如图3所示。考 虑到模拟起始阶段结构变化较大,我们选取20~60 ns 的模拟轨迹进行分析。对于起始结构,也就是 Aβ42 在PDB 中的结构,含有两段较长的 α螺旋(残基7~26 和28~40)。由图3可见,helix 的形成发生较明显变化。 300 K时,N端helix 形成的长度变短,C端helix 形 成的几率变低,大部分在60%以下,稳定性较差,且 长度变短;340 K时,在残基9~35 之间,形成 helix 几率均在 60%以上,起始态的两段 helix 又连接在一 起,形成一长helix;380 K 时,仅在残基 10~18 间存 在几率大于 60%的helix,其余残基形成 helix 几率较 低甚至不能形成 helix。在低温时,残基没有形成 strand,主要以 helix 或turn 的形式存在。在高温 380 K 时,除 helix 或turn 的形式,部分残基以低于 25%的 几率形成 strand。也就是说,在低温下,温度变化对 形成 strand 影响不大,而高温时形成 strand 的概率增 加。Aβ42 二级结构的变化情况表明了该蛋白的固有 无序特性,而且不同温度其不同区域形成二级结构概 率不一样,这启迪我们,温度和固有无序蛋白的无序 程度有联系,值得进一步深入研究。 为进一步分析不同温度下残基形成二级级结构 情况,我们利用STRIDE软件计算残基主链二面角, 进而得到残基主链二面角分布。二面角平均力势定义 为 F Copyright © 4 B i KT P 1611 16 21 26 31 36 41 46 0 20 40 60 80 ,其中 KB和T分别为波尔兹曼常 数和热力学温度,Pi为残基主链二面角落入区域 i的 几率。不同温度下残基主链二面角平均力势分布如图 4所示。不同区域定义如下[20],β折叠区域:–180˚ < φ < 0 v 且90˚ < Ψ < 180˚或–180˚ < Ψ < –120˚;α螺旋区 域:–180˚ < φ < 0˚且–120˚ < Ψ < 30˚。图中区域颜色越 深,对应该区域的平均力势越小,相应区域构象在模 拟中被采样的几率越大。由图 4可知,三种温度下蛋 白质各有一个平均力势极小区域,分别位于(–62.5˚, 100100 161116 21 26 3136 41 46 0 20 40 60 80 100 1 0611 16 21 26 31 36 41 46 20 40 60 80 Probablity/% Residue sequence Turn Bridge Helix Strand (b) Probablity/% Residue sequence Turn Turn Bridge Helix Strand (a) Probablity/% Residue se qu ence Bridge Helix Strand (c) Figure 3. The formation of secondary structure for residues during the simulation of 20~60 ns: (a) 300 K; (b) 340 K; (c) 380 K 图3. 20~60 ns 模拟区间残基形成二级结构的情况:(a) 300 K;(b) 340 K;(c) 380 K -180 -120-60060120180 -180 -120 -60 0 60 120 180 (a) -60 0 60 120 180 -180 -120-60060120180 -180 -120 -180 -120-60060120180 -180 -120 -60 0 60 120 180 (b) -10.00 -15.00 -14.00 -13.00 -12.00 -11.00 -9.000 -8.000 -7.000 -6.000 -5.000 -4.000 -3.000 -2.000 -1.000 Figure 4. Potential of mean forces obtained from Ramachandran (φ, ψ) angles distributions for the residues: (a) 300 K; (b) 340 K; (c) 380 K 图4. 残基 Ramachandran (φ, ψ)角的平均力势:(a) 300K;(b) 340 K;(c) 380 K (c)  基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化 42.5˚)、(–67.5˚, –67.5˚出的轨迹中,分别等间 – 于α螺旋区域。不同温度下力势极小值分别为 –14.74KJ·mol–1、–12.58 KJ·mol–1 和–11.22 KJ·mol–1。 与300 K、340 K 时二面角分布相比,在 380 K 时,残 基二面角位于 β折叠区域的概率增大,表明当温度 升 高到 380 K时,Aβ42 有从α螺旋向β折叠转换的趋势 。 3.3. 温度对三级结构的影响 温度变化导致二级结构的形成发生变化,同时也 会影响三级结构的稳定。为观察模拟过程中 Aβ42 构 象换转情况,我们以模拟时间为反映坐标,选取不同 时刻蛋白瞬时快照如图 5所示。在三种温度下,两螺 旋中的螺旋 2首先发生变换。在模拟最后时刻,蛋白 的L状天然拓扑结构完全消失。 在300 K 时,伴随着螺旋 2的变性,螺旋 1和螺 旋2间的夹角逐渐减小,最终螺旋 2由与螺旋 1由垂 直变为平行。同上述分析一致 ,Aβ42 在室温下并不 稳定(固有无序蛋白的特征)。在 340 K时,螺旋1和 螺旋 2间的夹角逐渐增大,由二者近似垂直,变为近 似在一条直线上,形成较长的一段螺旋,螺旋含量仍 维持在较高水平。380 K时,随着模拟时间的进行, 螺旋 2残基去螺旋化明显,同时与螺旋 1间夹角减小, 同300 K 一样两螺旋近似平行。与 300 K 不同的是, 380 K下螺旋含量较少,且 C段部分残基有形成β折 叠的趋势。 综上所述,无论是在室温还是高温下,Aβ42 蛋 白的三维结构均发生较大变化,原有L状拓扑结构消 失,两螺旋夹角变小,呈平形状(300 K和380 K),或 者形成一条直线(340 K)。 为了进一步确定体系采样构象的倾向类型,我们 对不同温度下模拟得到的构象系综进行聚类分析。构 象聚类基于构象之间骨架原子位置的均方根偏差 隔均匀选取 10,000 个构象作为分析对象,基于构象之 间的 RMSD进行聚类。不同温度下,含有构象数目最 大的三类如表 1所示。在300 K 时,轨迹中没有形成 具有绝对优势的构象。而在另外两种温度下,第 I类 含有 80%左右的结构。 不同温度下,每类的中心结构如图 6所示。300 K 时,类 I和II的中心结构中,两段螺旋呈平形排列, 残基螺旋含量减少。340 K时,蛋白成线状,螺旋含 量较高,与上述二级结构分析结果一致。在380 K 时, 类I中心结构中螺旋含量较低,二级结构和三级结构 均发生较大变化,部分残基形成 β折叠结构。也就是 说,高温可能导致β折叠结构的形成,进而容易聚集 形成淀粉样纤维。实验研究发现:在 AD 病患者中, 只有温度超过 373 K时,淀粉样纤维才能被分离出来[5]。 4. 结论 β-淀粉样多肽(β)聚集是阿尔茨海默病发病重要 因素。本文选取Aβ42 蛋白为对象,利用分子动力学 模拟的方法研究其在 300 K、340 K 和380 K 三种温度 下的构象变化。模拟采用 GROMACS软件包、 GROMOS 43A1 力场和充分含水模型,在三种温度下 分别对 Aβ42 进行了 60 ns 的独立分子动力学模拟。通 过对模拟轨迹分析发现:1) Aβ42在三种温度下,均 没有稳定结构,模拟一定时间后,其初始拓扑结构都 消失,说明该蛋白的固有无序特征;2) 温度对二级结 构和三级结构影响不同,在380 K时,L状天然拓扑 结构变为直线状,但二级结构含量仍较高;3) 低温时 蛋白质主要以α-helix 形式存在,而在高温(380 K)时, 有α-helix 向β-sheet转换的趋势。也就是说,高温可能 导致 Aβ42 的二级结构发生较大变化,加速蛋白质 )和(–57.5˚, –42.5˚)附近,均位 (RMSD)。在模拟最后 40 ns 输 Figure 5. The snapshot of protein during the simulations 图5. 模拟过程中蛋白瞬时快照 Copyright © 2013 Hanspub 5  基于分子动力学模拟研究温度致 Aβ42 蛋白构象变化 Table 1. The results of cluster 表1. 聚类结果 300 K 340 K 380 K I 47.73% 79.266% 0% 84. II 26.42% 12.97% 12.80% III 13.15% 4.70% 0.86% Figure 6. The central structure of each cluster 图6. 每类结构的中心构象 β-sheet 的形成,是形成聚集的基础,进而形成不溶性 纤维,这可为新的药物设计提供启迪;4) 不同温度对 Aβ42 的影响给我们很大启示,是否固有无序蛋白的 无序程度和温度存在某种联系,值得深入研究。 5. 致谢 感谢荷兰 Groningen 大学 Berendsen 教授提供的 GRO [1] 聚集性质比较[J]. 电子显微学报, 2003, 22(1): 21-25. [4] 于双妮, 刘立颖, 梁平作. β-淀粉样蛋白在阿尔茨海默病发病 和免疫治疗中的作用[J]. 中国病理学杂志, 2005, 34(2): 113- 114. s of America, 2005, 102(15): 5403- [8] 象转换的分子动力学模拟和结合自由能计算[J]. 物理化学学 报, 2012, 28(11): 2735-2744. cenzi, S. Tomaselli, R. Guerrini, et al. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar mi- [15] X. Daura, A. E. Mark Fetrization [17] H. J. C. Berendsen, J. P. M. . F. van Gunsteren, et al. [19] M. Heinig, D. Fran. STRIDE: A web server fory 段 的分子动力学模拟[J]. 物理化学学报, 2012, 28 (7): 1665-1675. MACS 软件。 参考文献 (References) 张伟, 刘志敏. 阿尔茨海默氏症的研究进展[J]. 中国生物工 程杂志, 2003, 23(12): 13-16. [2] R. Nelson, M. R. Sawaya, et al. Structure of the cross-β spine of amyloid-like fibrils. Nature, 2005, 435(7043): 773-778. [3] 武一, 吉尚戎, 蒋伍玲等. β淀粉样蛋白 Aβ1-40和Aβ1-42 的 [5] T. Takeda, D. K. Klimov. Temperature-induced dissociation of Aβ monomers from amyloid fibril. Biophysical Journal, 2008, 95(4): 1758-1772. [6] Z. X. Cao, L. Liu, L. L. Zhao, et al. Effects of different force fields and temperatures on the structural character of abeta (12 - 28) peptide in aqueous solution. International Journal of Mo- lecular Sciences, 2011, 12(11): 8259-8274. [7] Y. Xu, J. Shen, X. Luo, et al. Conformational transition of amy- loid beta-peptide. Proceedings of the National Academy of Sci- ences of the United State 5407. G. Wei, A. I. Jewett and J. E. Shea. Structural diversity of dimers of the Alzheimer amyloid-beta (25 - 35) peptide and polymor- phism of the resulting fibrils. Physical Chemistry Chemical Physics, 2010, 12(14): 3622-3629. [9] D. J. Selkoe. Ahheimer’s disease: Genes, proteins and therapy. Physiological Reviews, 2001, 81(2): 741-766. [10] K. A. Coalier, G. S. Paranjape, S. Karki, et al. Stability of early-stage amyloid-β(1-42) aggregation species. Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics, 2012, 1834(1): 65-70. [11] T. Zhao, Y. Zeng and A. R. Kermode. A plant cell-based system that predicts Aβ42 misfolding: Potential as a drug discovery tool for Alzheimer’s disease. Molecular Genetics and Metabolism, 2012, 107(3): 571-579. [12] 董晓燕, 都文婕, 刘夫锋. 多肽抑制剂抑制淀粉质多肽 42 构 [13] O. Cres croenvironment. Similarity with a virus fusion domain. Euro- pean Journal of Biochemistry, 2002, 269(22): 5642-5648. [14] D. van der Spoel, R. van Drunen and H. J. C. Berendsen. Gron- ingen machine for chemical simulations. Groningen: Department of Biophysical Chemistry, BIOSON Research Institute, 1994. and W.. van Gunsteren. Param of aliphatic CHn united atoms of GROMOS96 force field. Jour- nal of Computational Chemistry, 1998, 19: 535-547. [16] J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. Journal of Com- putational Physics, 1977, 23(3): 327-341. Postma, W Molecular dynamics with coupling to an external bath. Journal of Computational Physics, 1984, 81(8): 3684-3690. [18] S. H. Chong, S. Ham. Impact of chemical heterogeneity on protein self-assembly in water. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(20): 7636-7641. ishm secondar structure assignment from known atomic coordinates of proteins. Nucleic Acids Research, 2004, 32(Web Server Issue): W500- W502. [20] 许朝莹, 赵立岭, 曹赞霞等. 残基突变对 P53-DNA 结合域肽 构象影响 Copyright © 2013 Hanspub 6 |