 Applied Physics 应用物理, 2013, 3, 1-5 http://dx.doi.org/10.12677/app.2013.31001 Published Online January 2013 (http://www.hanspub.org/journal/app.html) Analysis of Self-Diffusion of FCC Transition Metals by Modified Analytical Embedded-Atom Method* Guoxiang Chen#, Xiaoli Li School of Science, Xi’an Shiyou University, Xi’an Email: #guoxchen@xsyu.edu.cn Received: Nov. 17th, 2012; revised: Nov. 29th, 2012; accepted: Dec. 12th, 2012 Abstract: The energy during the process of self-diffusion in FCC transition metals Ni, Pd, Pt, Cu, Ag, Au and Al has been calculated by using modified analytic embedded-atom method (MAEAM). For each kind of two diffusion mecha- nisms nearest-neighbor (NN) and next-nearest-neighbor (NNN), the energy curve is symmetric and the maximum value of the energy appears at the middle point of the diffusion path. Determined mono-vacancy formation energy 1 f v E, mi- gration energy and activation energy for self-diffusion agree well with available experimental data of NN diffusion and are better than those obtained by the embedded-atom method (EAM). Compared the energies corre- sponding to two diffusion mechanisms, the NN diffusion needs the lowest activation energy (and thus the lowest migra- tion energy). So that, the NN mono-vacancy diffusion is favorable in FCC transition metals. 1 m v E1v Q Keywords: Self-Diffusion; FCC Transition Metals; Vacancy 面心立方过渡金属自扩散的改进分析型嵌入原子法分析* 陈国祥#,李晓莉 西安石油大学理学院,西安 Email: #guoxchen@xsyu.edu.cn 收稿日期:2012 年11月17 日;修回日期:2012 年11月29 日;录用日期:2012 年12 月12日 摘 要:采用改进分析型嵌入原子法(MAEAM)计算了面心立方过渡金属 Ni、Pd、Pt、Cu、Ag、Au 和Al 自扩 散过程中的能量。对于最近邻(NN)和次近邻(NNN) 二种扩散机制,其能量曲线均为对称曲线且能量的最大值均 位于各自扩散路径的中点。计算得到的单空位形成能 1 f v E、迁移能 和自扩散激活能 比用嵌入原子法(EAM) 计算的结果更接近 NN扩散的实验数据。计算结果表明NN 扩散的激活能最低(迁移能也为最低),因此面心立方 过渡金属中的最可几扩散为单空位最近邻扩散。 1 m v E1v Q 关键词:自扩散;面心立方过渡金属;空位 1. 引言 早在 1984 年,Finnis 等人[1]用F-S 模型计算了体 心立方(BCC)过渡金属的空位形成能,迁移能和自扩 散激活能。1992年,Guellil 等人[2]用分析型嵌入原子 法(AEAM)也计算了自扩散过程中的这些值。但是他 们都没有提供空位在 BCC 过渡金属中的自扩散过程。 另外张邦维等人[3]用AEAM 模型研究过渡金属二元 合金的稀溶解热与形成焓,随后又调整势函数与嵌入 函数以改进计算结果与实验结果的符合程度,改进后 的模型在处理过渡族 BCC 二元合金的稀溶解热与形 成焓方面是成功的。但改进的模型依然不能处理Cr *资助信息:陕西省教育厅科研计划项目(批准号:11JK0822)资助的 课题。 #通讯作者。 Copyright © 2013 Hanspub 1  面心立方过渡金属自扩散的改进分析型嵌入原子法分析 等负柯西压元素的问题,这限制了方法的应用,特别 是对体心立方金属和金刚石结构材料,因此张邦维建 立了一个改进分析型嵌入原子法(MAEAM)[4,5]。我们 前期论文采用 MAEAM成功的计算了七种 BCC 过渡 金属(Fe、W、Mo、Cr、Ta、Nb 和V)体内单空位形 成能,迁移能以及自扩散激活能[6]。本文以Ni、Pd、 Pt、Cu、Ag、Au 和Al七种面心立方(FCC)过渡金属 为例,采用 MAEAM 计算七种过渡金属体内单空位形 成能,迁移能和自扩散激活能。从能量最小化观点, 讨论了在 FCC 过渡金属中最可几的扩散即空位或原 子在迁移过程中激活能最低的扩散。本文的计算结果 相比其他模型的计算值,要与实验数据更接近些,这 表明 MAEAM 能更有效的模拟 FCC 过渡金属自扩散。 2. 计算模型 在MAEAM 模型中,系统的总能量 表示为[4] total E total 1 2 iij iijii EF rM i P ij , (1) i ji f r , (2) 2 i ji Pf i j r . (3) 其中 i F 是在除第 个原子外的其他原子组成基体 中再嵌入第 i个原子的嵌入能,它仅是其他原子在第 i i 个原子所在处产生的背景电子密度 ii ji j f r 的 函数; ij f r 度; 是第个原子在第个原子所在处产生的ji 电子密 ij r是第 i个原子和第 j个原子间的距离; ij r 是第 i原子和第j个原子间的相互作用势; 个 i M P 所引 是修正项,表示原子电子密度非球型对称分布 系统总能量的变化。嵌入函数 i F 起的 、原子 间的相互作用势 ij r 、修正项 i M P和 密度函 数 ij 电子 f r采用如下 [3,4] 形式 0 n ii eie FF1lnn , (4) 2412 01 12131 ijij eij eeij rkkrr krr krr , (5) 2 1exp 1 ii iee PP MP PP 2 , (6) 6 1 ijee ij frf rr. (7) 其中下标 表示平衡 态[4] e状态, 1e r是平衡状态下第一近邻 的距离。本文选用的平衡状的电子密度为 35 1ecf fEE (8) 其中 34a 为FCC 金属中每个原 (4)~ 子所占的体积。 公式 (6)中的七个参数 n、0 F 、 、0 k、1 k、 2 k和 形成能 3 k可根据所考虑FCC 金属的结能 c E,单空 1 合位 f E,晶 格常 数a,弹性常数 11 C、和 44 C由 下列各式 得 12 C 求 111211 12 144 2 216 f CCCC nEC , (9) 01cf F EE , (10) 2 1 C nF , (11) 12 44 0 1 32 8 C 01 44 1211 1 9 15481 29892989 42840 f kE CCC (12) 14412 11311 939 939 9520 kCC 11 C , (13) 24412 133 32 32 1020 kCC 11 C , (14) 34412 89 5355 kCC 11 C . (15) 根据张邦维等[3]的分析,(5)式表示的相互作用势 ij r 于距离大 只适用于原子间距小于第二近邻2e r的情况,对 于2e r时,可使用一个三次条样作为截尾 函数 函数 23 01 23 22 2 11 ij ijij ij ee e rr r rll ll rr r 1 2eijc rrr 6) 其中四个参数、、 、和截尾距离 分别 7) 12 (1 0 l1 l2 l3 lc r为 24 12 2 3 lkk sks , (1 00 1 sk 24 11 23 2412lksks ks , (18) 0 1 22 3 2 11 l l l , (19) 0 1 323 2 11 l l l , (20) Copyright © 2013 Hanspub 2  面心立方过渡金属自扩散的改进分析型嵌入原子法分析 2e . (21) 23 0.75 cee rr rr 其中 和分别为平衡状态下第二近邻和第三近邻 2e r 的距离, 3e r 21ee s rr,2ce rr 。 在绝对温度为零度以上的任何温度下,晶体的晶 格中的紊乱分布可 以使 表1 应的计算单元,在计算单元的周围 3. 自扩散过程中能量计算 格中总会存在一些空位,空位在晶 熵增加。如果一个原子落在空位旁边,它就可能 跳进空位中,如图 1所示,导致原子原来的位置变成 空位,另外邻近原子也可能占据这个新形成的空位, 使空位继续运动,这就是空位扩散机制。在本文中通 过移除完整晶体晶格中的一个原子的位置而形成单 空位,考虑到单空位朝它邻近原子的迁移过程等价于 邻近原子向空位移动。图 2示意单空位的不同扩散机 制,即最近邻(NN)、次近邻(NNN)二种扩散机制。 有关 Ni、Pd、Pt、Cu、Ag、Au和Al 七种 FCC 过渡金属的输入参数和MAEAM 模型参数分别列在 和表 2中。 在完整的立方金属晶格中,在计算中对于空位所 在位置的不同取相 Figure 1. Vacancy diffusion mechanism 图1. 空位扩散机制 (a) (b) Figure 2. Two different migratechanisms of mono-vacancy: (a) Nearest-neighbor (NN); (brest-neighbor (NNN). e solid circles repent face atoms and blank squares represent vacancy ion m ) Next-neaTh resent top atoms, blank circles repres 图2. 单空位在面心立方过渡金属中的二种不同扩散机制:(a) 最 近邻;(b) 次近邻。实心、空心圆环和空心方块分别代表顶、面上 的原子以及空位 界条件。通过从 层上移走一个采用固定边n原子到无 穷,从而在计算单元中形成空位结构,单空位形成能 1 J v fv vt tc EEEE (22) v和是体系中 E可以通过下式得到 : 式中 存在空位和没有空位时的总能 量。 ,弥补因原子数目 的变化。 计算得到七种FCC 过渡金属的形成能如表 3 计算结果与实验相符的。 参数 a (nm) (eV) [7] t E c E t E 是结合能 的不同引入的能量 所示,比较实验数据和其他模型的计算结果,本文的 很好 Table 1. Input physical parameters of seven-FCC transition metals 表1. 七种 FCC 过渡金属的输入 参数 c1 E f v E(eV) (GPa) (GPa)(GPa) 11 12 44 CC C Ni0.352364.44 1.45 1540 960 760 Pd0.389073.89 1.30 1400 1080 450 51215Pt0.39239.84 .20 120 720 20 Cu0.361473.49 1.17 1050 760 470 Ag0.408572.95 1.10 770 570 280 Au0.407883.81 0.90 1190 1010 260 Al0.404963.39 0.64 650 460 270 T 2M l par of-FCsi t i onls 七种过渡金属的 MAEAM模型参数 参数 Ni Pd Pt Cu Ag Au Al able. MAEAmodeameters sevenC tran meta 表2. FCC n 0.304 0.364 0.499 0.272 0.313 0.440 0.364 e f 0.9 0.0.493 0.34 0.347 0.3404535377 0.26 0 F (eV) 2.24.2 12.2990.590640.320.850 910.750 0.034 0.247 0.423 0.086 0.132 0.327 0.053 0 k(e (e ) V) −0.782 −0.663 −0.717 −0.601 −0.495 −0.451 −0.425 1 k(eV) 0.519 0.448 0.486 0.426 0.321 0.306 0.306 2 kV−0.070 −0.067 −0.065 −0.072 −0.047 −0.047 −0.046 3 k(eV) 0.102 0.082 0.097 0.070 0.059 0.055 0.056 ueo experim 表文的成 数 值 eV) Table3. Calclated th vacanc tica y formation energy and ther ental dat 计算 a and theor 空位形 e 能及其 l calculations (in u 他实验 nits of e 论计算 V) (单位3. 本 据和理 金属 Present work Exp.data EAM[10] Ni 1.664 1.70[8] 1.71 Pd 1.534 1.54[9] 1.58 Pt 1.580 1.60[9] 1.77 Cu 1.284 1.31[8] 1.33 Ag 1.118 1.16[8] 0.97 Au 0.929 0.92[8] 1.04 Al 0.659 0.67[8] - Copyright © 2013 Hanspub 3 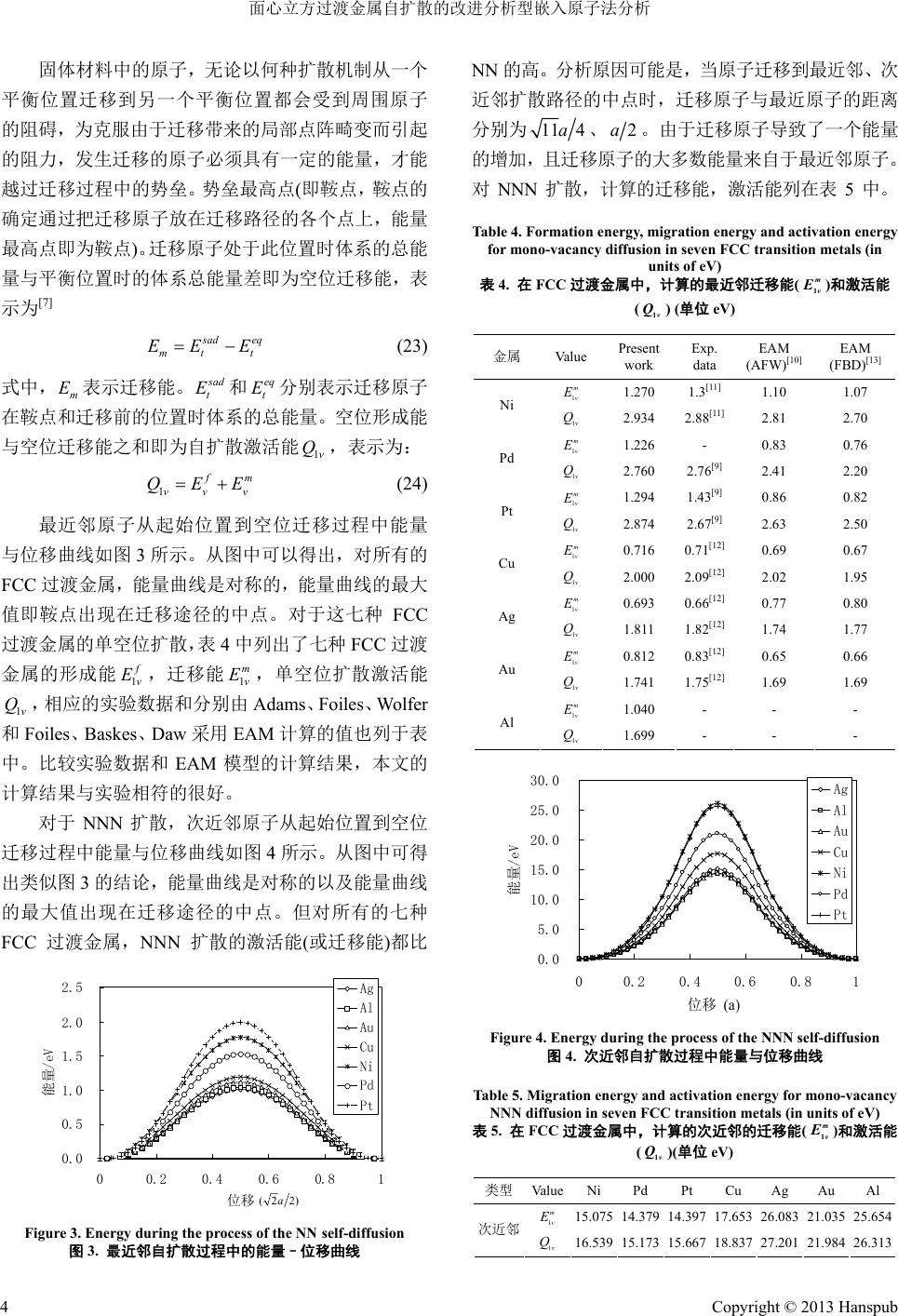 面心立方过渡金属自扩散的改进分析型嵌入原子法分析 固体材料中的 ,无论以 扩散机制个 平衡位置迁移到另一个平衡位置都会受到周围原子 的阻碍 为克服由于迁移带来的局部点阵畸变而引起 的阻力 原子 何种从一 , ,发生迁移的原子必须具有一定的能量,才能 越过迁移过程中的势垒。势垒最高点(即鞍点,鞍点的 确定通过把迁移原子放在迁移路径的各个点上,能量 最高点即为鞍点)。迁移原子处于此位置时体系的总能 量与平衡位置时的体系总能量差即为空位迁移能,表 示为[7] s ad eq mt t EE E (23) 式中, m E表示迁移能。 s ad t E 时体 自扩 和 分别表示迁移原子 在鞍点和迁移前的位置系 与空位迁移能之和即为 ,表示为: eq t E 的总 散激活能 能量。空位形成能 1v 1 Q f m vvv QEE (24) 最近邻原子从起始位置到空位迁 过程中能量 移 与位移曲线如图 3所示。从图中 FCC 过渡金属,能量曲线是对称的,能量曲线的最大 值即鞍点出 可以得出,对所有的 现在迁移途径的中点。对于这七种FCC 过渡金属的单空位扩散,表 4中列出了七种 FCC 过渡 金属的形成能 1 f v E,迁移能 1 m v E,单空位扩散激活能 1v Q,相应的实验数据和分别由Adams、Foiles、Wolfer 和Foiles、Baskes、Daw 采用 EAM计算的值也列于表 中。比较实验数据和EAM模型的计算结果,本文的 与实验相符的很好。 对于 NNN 扩散,次近邻原子从起始位置到空位 迁移过程中能量与位曲线如图 4所示。从图中可得 出类似图 3的结论,能量曲线是对称 计算结果 移 的以及能量曲线 的最大值出现在迁移途径的中点。但对所有的七种 FCC 过渡金属,NNN扩散的激活能(或迁移能)都比 0.0 0.5 1.0 1.5 2.0 2.5 00.20.4 0.6 0.81 位移 能量/eV Ag Al Au Cu Ni Pd Pt )22(a Figure 3. Energy during the process of the NN self-diffusion 图3. 最近邻自扩散过程中的能量–位移曲线 NN 的高。分析原因可能是,当原子迁移 到最近邻、次 近邻扩散路径的中点时,迁移原子与最近原子的距离 分别为 114a、2a。由于迁移原子导致了一个能量 的增加,且迁移原子的大多数能量来 自于最近邻原子。 对NNN 扩散,计算的迁移能,激活能列在表 5中。 Table 4. Formation energy, migration energy and activation energy for mono-vacancy diffusion in seven FCC transition metals (in units of eV) 表4. 在FCC过渡金属中,计算的最近邻迁移能(m v E 1)和激活能 (v Q1) (单位 eV) 金属 Va lu e Present work Exp. data EAM (AFW)[10] EAM (FBD)[13] 1 m v E 1.270 1.3[11] 1.10 1.07 Ni 34 2.88[11] 2.81 2.70 1 m v E 1.226 - 0.83 0.76 Pd 1v Q 2.760 2.76[9] 2.41 2.20 1v Q 2.9 1.294 1.43[9] 0.86 0.82 0.71 69 0.67 0.69 0.77 0.80 Ag 1 0 Au Al 1 Pt 1v Q 2.874 2.67[9] 2.63 2.50 6 0.71[12] 0. m v E 1 m v E Cu 1v Q 2.000 2.09[12] 2.02 1.95 3 0.66[12] 1 m v E 1v Q 1.811 0.812 .82[12] .83[12] 1.74 0.65 1.77 0.66 1 m v E 1v Q 1.741 1.75[12] 1.69 1.69 1 m v E 1.040 - - - 1v Q 1.699 - - - 0.0 5.0 10.0 15.0 20.0 25.0 30.0 00.2 0.40.6 0.81 能量/e A V g Al Au Cu Ni Pd Pt 位移 (a) Figure 4. Energy during the process of the NNN self-diffusion 图4. 次近邻自扩散过程中能量与位移曲线 Table 5. Migration energy and activation energy for mono-vacancy NNN diffusion in seven FCC transition metals (in units of eV) 表5. 在FCC过渡金属中,计算的次近邻的迁移能(m v E 1)和激活能 )(单位 eV) 类型 ValueNi Pd Pt Cu Ag Au Al (v Q1 1 m v E15.075 14.379 14.397 17.653 26.083 21.035 25.654 次近邻 16.539 15.173 15.667 18.837 27.201 21.984 26.313 1v Q Copyright © 2013 Hanspub 4  面心立方过渡金属自扩散的改进分析型嵌入原子法分析 Copyright © 2013 Hanspub 5 通过比较表 4可以得出,对于FCC 过渡金属中的二种 扩散机制,最低的激活能(或迁移能)对应着NN扩散, 这意味着在 FCC 过渡金属中的最可几扩散为单空位 NN 扩散。 4. 结论 本文采用 MAEAM 计算了面心立方过渡金属 Ni、 Pd、Pt、Cu、Ag、Au 和Al 自扩散过程中的能量。 于 曲线且能 点 对 NN 和NNN 两种扩散机制,其能量曲线均为对称 量的最大值均位于各自扩散路径的中 。所 确定的单空位形成能 1 f v E﹑迁移能 1v E和自扩散激活 Q比EAM 计算的结果更接近NN 扩散的实验数 m 能 据。 对应于 NN扩散 激(迁移能也为最 因此,面心立方过渡金属中的最可几扩散为单空位最 近邻扩散。 5. 致谢 (References) [1] uellil, J. B. Adams. The application of the analytic em- bedded atom method to bcc metals and alloys. Journal of Mate- [3] B. W. Zhang, Y. F. Ouyang. Theoretical calculation of view B, 1993, 48(5): 3022-3029. Zhang, X. L. Shu and B. Y. Huang. Calculation of formation enthalpies and phase stability for Ru-Al alloys us- ing an analytic embedded atom model. Journal of Alloys and -162. uang, F. Gao and D. J. Bacon. Analytic modified embedded atom potentials for HCP metals. n metals calculated with MAEAM. Physica B, 2007, tion in hcp metals. Journal olfer. Self-diffusion and . Y. Hu, X. L. Shu. Theory of embedded Embedded-atom- 7983-7991. 45-55. [2] A. M. G rials Research, 1992, 7(3): 639-652. thermo- dynamic data for bcc binary alloys with the embedded-atom me- thod. Physical Re [4] W. Y. Hu, B. W. Compounds, 1999, 287(1-2): 159 [5] W. Y. Hu, B. W. Zhang, B. Y. H Journal of Physics: Condensed Matter, 2001, 13(6): 1193-1213. [6] J. M. Zhang, G. X. Chen and K. W. Xu. Self-diffusion of BCC transitio 390(1-2): 320-324. [7] M. I. Pascuet, R. C. Pasianot and A. M. Monti. Computer simu- lation of surface-point defects interac 1v 的 活能最低低)。[9] B. T. A. Mckee, W. Trifthauser and A. T. Stewart. Vacancy-for- mation energies in metals from positron annihilation. Physical Review Letters, 1972, 28: 358-360. [10] J. B. Adams, S. M. Foiles and W. G. W of Molecular Catalysis A, 2001, 167: 165-170. [8] W. Schüle, R. Scholz. On point defect and interactions in metals. Tokyo: University of Tokyo Press, 1982: 257. impurity diffusion of free metals using the five-frequency model and the embedded atom method. Journal of Materials Research, 1989, 4(1): 102-112. [11] N. L. Peterson. Isotope effect in self-diffusion in palladium. Physical Review, 1964, 136: A568-A574. [12] B. W. Zhang and W 本文感谢陕西省教育厅科研计划项目(11JK0822) 的支持;感谢西安石油大学博士科研启动项目(2011 BS024)的支持。 atom method and its application to material science. Changsha: Hu’nan University Press, 2003: 84-85. [13] S. N. Foiles, M. I. Baskes and M. S. Daw. method functions for the FCC metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys. Physical Review B, 1986, 33(12): 参考文献 M. W. Finnis, J. E. Sinclair. A simple empirical N-body potential for transition metals. Philosophical Magazine A, 1984, 50(1): |